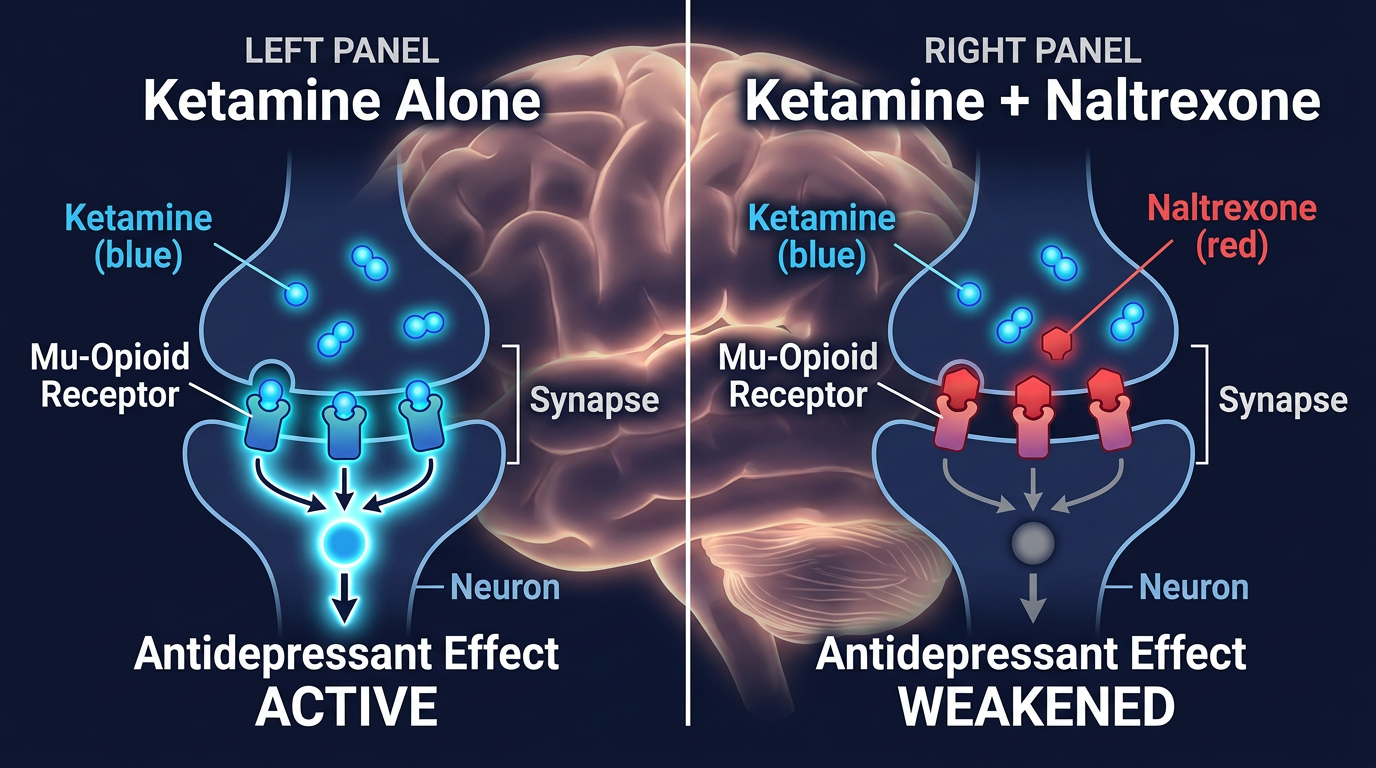
Opioid System Activation is Required for Ketamine’s Antidepressant Effects
Ketamine is fast—a single subanesthetic infusion reduces severe depression symptoms within hours, not weeks. But the mechanism remains incompletely understood. For nearly two decades, research assumed glutamate flooding the brain explained everything: ketamine blocks NMDA receptors on inhibitory neurons, releasing the brakes on glutamate release, which then triggers neuroplasticity and mood recovery. A new double-blind crossover study reveals this picture is incomplete. When researchers blocked opioid receptors with naltrexone before ketamine infusion, depression improved 28% less and the brain’s glutamate surge shrank by 28%. The opioid system is not a minor player—it is essential for ketamine’s antidepressant power.
Key Findings
- Naltrexone pretreatment reduced ketamine-induced glutamate surge by 28% in the anterior cingulate cortex (F₁,₂₅₃ = 4.83, P = 0.029; Cohen’s d = 0.34)
- Depression symptom reduction on day 1 post-infusion dropped significantly with naltrexone: placebo+ketamine reduced MADRS by 14.65 points vs. naltrexone+ketamine by 10.50 points (P = 0.023, Cohen’s d = 0.60)
- Blocking mu opioid receptors weakened but did not eliminate ketamine’s antidepressant effects, indicating the opioid system is necessary but not sufficient
- Dissociative and psychotomimetic effects remained unchanged by naltrexone, revealing a separate neurobiological mechanism for ketamine’s hallucinogenic side effects
- Male participants showed larger naltrexone-induced reductions in ketamine response than females, suggesting sex-specific opioid-dependent effects
- Blinding was verified successful (James Blinding Index = 0.46–0.38), confirming rigorous double-blind methodology
Source: Nature Medicine (2025) | Jelen et al.
Real-Time Brain Imaging Reveals Opioid-Glutamate Synergy During Ketamine Infusion
The study enrolled 26 adults with major depressive disorder (mean age 35.08 years, 50% female, mean HAM-D = 21.65, indicating moderate-to-severe baseline depression). Participants had failed 1–10 prior antidepressants, with 46% currently taking antidepressant medication (continued throughout the study). All 26 completed both arms of a randomized, double-blind, placebo-controlled crossover design, with an average 19-day interval between visits.
On each visit, participants received either 50 mg oral naltrexone or placebo, followed 1 hour later by an IV ketamine infusion (0.5 mg/kg over 40 minutes). Critically, brain glutamate activity was measured in real time using functional proton magnetic resonance spectroscopy (¹H-fMRS), capturing the neurochemical dynamics during the first 30 minutes of infusion—the period of peak glutamate surge. The researchers focused on the anterior cingulate cortex, the brain region most involved in emotional processing and where ketamine’s glutamate effects are strongest.
Glutamate Surge Magnitude Tracks with Opioid Receptor Activity
Both naltrexone and placebo conditions produced glutamate elevation during ketamine infusion, consistent with prior research. Peak changes occurred 15–25 minutes into the infusion, and the pattern was similar across conditions. However, the magnitude of the surge differed sharply (F₁,₂₅₃ = 4.83, P = 0.029). Mean peak glutamate + glutamine to total N-acetylaspartate ratio (Glx/tNAA) in the placebo condition reached +0.055; in the naltrexone condition, it plateaued at +0.038—a 28% reduction. This difference remained consistent across the entire 30-minute infusion window, suggesting that opioid receptor activity directly modulates glutamatergic response to ketamine.
The researchers performed sensitivity analyses adjusting for age, sex, brain white matter and gray matter composition, ¹H-fMRS signal quality, concurrent antidepressant use, and visit order. All models confirmed the same result: blocking mu opioid receptors with naltrexone attenuates ketamine-induced glutamate elevation. This robustness suggests the opioid-glutamate interaction is a fundamental feature of ketamine’s neurochemistry, not a chance finding.
Attenuated Glutamate Surge Predicts Reduced Depression Response
The dampened glutamate surge had behavioral consequences. Depression severity was assessed using the Montgomery–Åsberg Depression Rating Scale (MADRS), the gold standard clinician-rated measure in antidepressant research. At baseline, both conditions were equivalent (placebo: mean = 28.73, SD = 5.75; naltrexone: mean = 27.23, SD = 5.54, p > 0.05).
On day 1 post-infusion, the difference emerged clearly:
- Placebo + ketamine: MADRS reduction of −14.65 points (SD 7.77)
- Naltrexone + ketamine: MADRS reduction of −10.50 points (SD 5.91)
- Difference: 4.15 points (condition-by-time interaction, F₁,₇₄ = 5.39, P = 0.023; Cohen’s d = 0.60)
This represents a 28% reduction in antidepressant benefit with naltrexone pretreatment—a medium effect size with clear clinical relevance. On a scale where a 4–5 point shift determines the difference between remission and partial response, the clinical significance is substantial. By day 3 and day 7, the gap narrowed, suggesting either opioid-independent mechanisms engaged over time or recovery of opioid system signaling. Self-report depression measures (QIDS-SR) and anhedonia scales (SHAPS, TEPS) trended in the same direction as the primary MADRS finding but did not reach statistical significance, though this may reflect reduced statistical power in secondary outcomes.
Dissociative and Psychotomimetic Effects Persist Despite Opioid Blockade
A critical finding was what naltrexone did not affect: ketamine’s hallucinogenic side effects. The Clinician Administered Dissociative States Scale (CADSS) showed no significant difference between placebo+ketamine (mean = 31.62, SD = 13.23) and naltrexone+ketamine (mean = 31.77, SD = 12.62; F₁,₂₅ = 0.003, P = 0.959). Similarly, all subscales of the self-report Psychotomimetic States Inventory (PSI) were unchanged by naltrexone. This dissociation between effects—opioid-dependent mood benefit but opioid-independent dissociation—is as informative as a positive finding. It indicates that ketamine’s hallucinogenic properties arise through a separate mechanism, likely direct NMDA antagonism or downstream glutamate signaling in sensory cortices rather than through mu opioid receptors.
Proposed Mechanisms: How Opioid Receptors Amplify Glutamate Release
The exact mechanism by which opioid activation enhances ketamine-induced glutamate surge remains to be fully elucidated, but two models are biologically plausible. The first involves local GABAergic inhibition in the anterior cingulate cortex. Mu opioid receptors are abundantly expressed on GABAergic interneurons. Under normal conditions, endogenous opioids (endorphins, enkephalins) activate these receptors, suppressing GABA release and reducing inhibition on pyramidal neurons. Naltrexone blocks these receptors, potentially strengthening GABAergic tone and capping pyramidal glutamate output even when NMDA antagonism attempts to disinhibit them.
A second mechanism involves direct opioid-glutamate interactions. Recent evidence suggests that ketamine itself acts as a weak mu opioid agonist at physiologically relevant concentrations, and may allosterically enhance mu opioid receptor signaling. Naltrexone directly blocks these mu opioid responses, preventing the amplification of glutamatergic neurotransmission. Both mechanisms could operate simultaneously, or involve interactions with other opioid receptor subtypes (delta, kappa) that also modulate glutamate release. A direct pharmacokinetic interaction is unlikely—ketamine undergoes metabolism via CYP2B6 and CYP3A4, while naltrexone uses different enzymatic pathways, making drug-drug interactions at the metabolic level improbable.
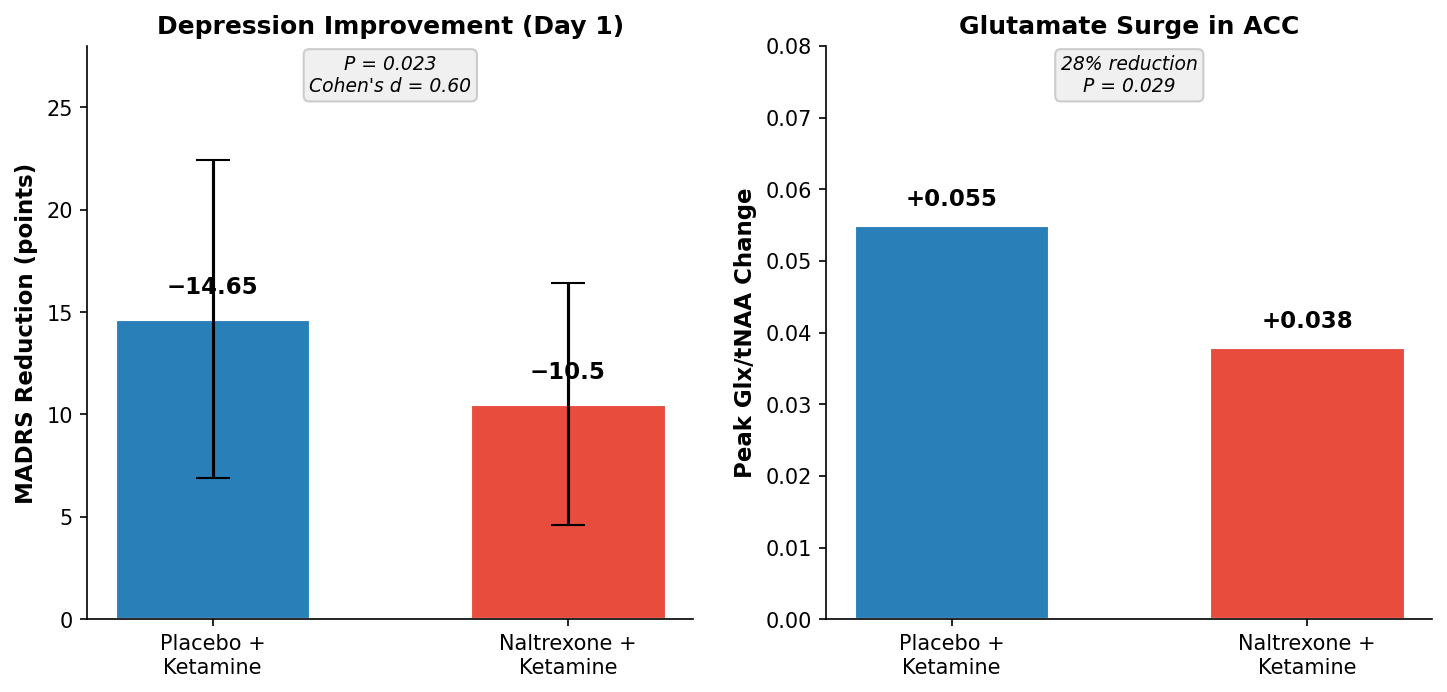
Image file: naltrexone-ketamine-depression.png — Day-1 MADRS symptom reduction and peak glutamate surge (Glx/tNAA) comparing placebo+ketamine vs. naltrexone+ketamine conditions (N = 26 completers)
Clinical Implications for Patients and Practitioners
If you take naltrexone for alcohol or opioid use disorder: If you are currently taking naltrexone and your psychiatrist is considering ketamine for treatment-resistant depression, this study reveals an important interaction. Naltrexone reduces ketamine’s antidepressant benefit by approximately 28% and dampens the brain’s glutamate response. This does not rule out ketamine as a treatment option, but it suggests the need for careful dose optimization, longer observation periods to capture delayed antidepressant response, or a discussion with your clinician about whether temporarily discontinuing naltrexone during a supervised ketamine trial might be feasible and safe. The answer is highly individual and depends on your addiction history and relapse risk.
Ketamine’s mechanism is not “just glutamate”: This work definitively closes the chapter on simplistic mechanistic models. Ketamine is not “just a glutamate drug” any more than SSRIs are “just serotonin drugs.” Both systems require intact signaling through multiple neurobiological pathways for therapeutic benefit. The opioid system is an essential collaborator in ketamine’s mood effects, not a peripheral player. Future antidepressant development should deliberately target both systems to amplify efficacy or enable lower doses with fewer side effects.
Combining opioid and glutamate systems for enhanced antidepressant development: Preclinical evidence and this clinical study suggest that agents combining opioid-system-enhancing effects (selective mu opioid agonists, endorphin-boosting protocols, or compounds with partial mu agonist activity) with ketamine or other glutamate-modulating agents could potentially enhance antidepressant efficacy or permit lower ketamine doses. Sex-specific responses were noted—males showed numerically larger naltrexone-induced reductions in ketamine response than females—warranting future investigation and potentially enabling personalized medicine approaches to treatment selection.
The dissociation between mood and hallucinogenic effects suggests separate targets: Since naltrexone does not reduce dissociation or psychotomimetic effects, blocking the opioid system alone will not eliminate ketamine’s hallucinogenic side effects. Future research should target other mechanisms (NMDA antagonism in sensory circuits, alpha-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid receptor signaling, or connectivity in perceptual cortices) if the goal is to preserve antidepressant benefit while eliminating disorienting side effects.
Study Strengths and Limitations
Strengths include rigorous double-blind randomized crossover design with verified blinding (James Blinding Index = 0.46–0.38, indicating successful masking). The use of functional ¹H-fMRS to measure real-time glutamatergic dynamics during drug administration is novel for studying opioid-ketamine interactions. The sample size (n = 26 completers) exceeds prior opioid-antagonism trials. All participants were carefully characterized with validated depression severity measures and completed comprehensive assessments of dissociation and psychotomimetic effects.
Limitations include the absence of a placebo-infusion control condition, preventing determination of whether naltrexone affects baseline glutamate independent of ketamine. The ¹H-fMRS measure combines glutamate and glutamine (Glx) rather than measuring each separately, reducing neurochemical specificity. The sample had moderate treatment resistance (1–2 prior antidepressant failures), potentially limiting generalizability to ultra-treatment-resistant cases. Racemic ketamine was used; (S)-ketamine has higher mu opioid affinity, so enantiomer-specific effects remain unknown. The sample size was insufficient for adequately powered sex-difference analysis, though trends suggest males may show larger opioid-dependent effects. The 19-day interval between visits was brief; partial depression relapse by visit 2 may have reduced statistical power for self-report mood measures.
Citation: Jelen, L. A., Lythgoe, D. J., Stone, J. M., Young, A. H., & Mehta, M. A. (2025). Effect of naltrexone pretreatment on ketamine-induced glutamatergic activity and symptoms of depression: a randomized crossover study. Nature Medicine, 31, 2958–2966. doi:10.1038/s41591-025-03800-w
Authors’ affiliations: Institute of Psychiatry, Psychology and Neuroscience, King’s College London; South London and Maudsley NHS Foundation Trust; Brighton and Sussex Medical School, University of Sussex; Imperial College London.