The Broken Repair Crew: How TDP43 Sabotages Genome Stability
TL;DR: TDP43, the protein that misfires in ALS and FTD, secretly controls DNA repair genes—and when it breaks, mutations pile up in neurons, potentially explaining both neurodegeneration and the cancer link in these diseases.
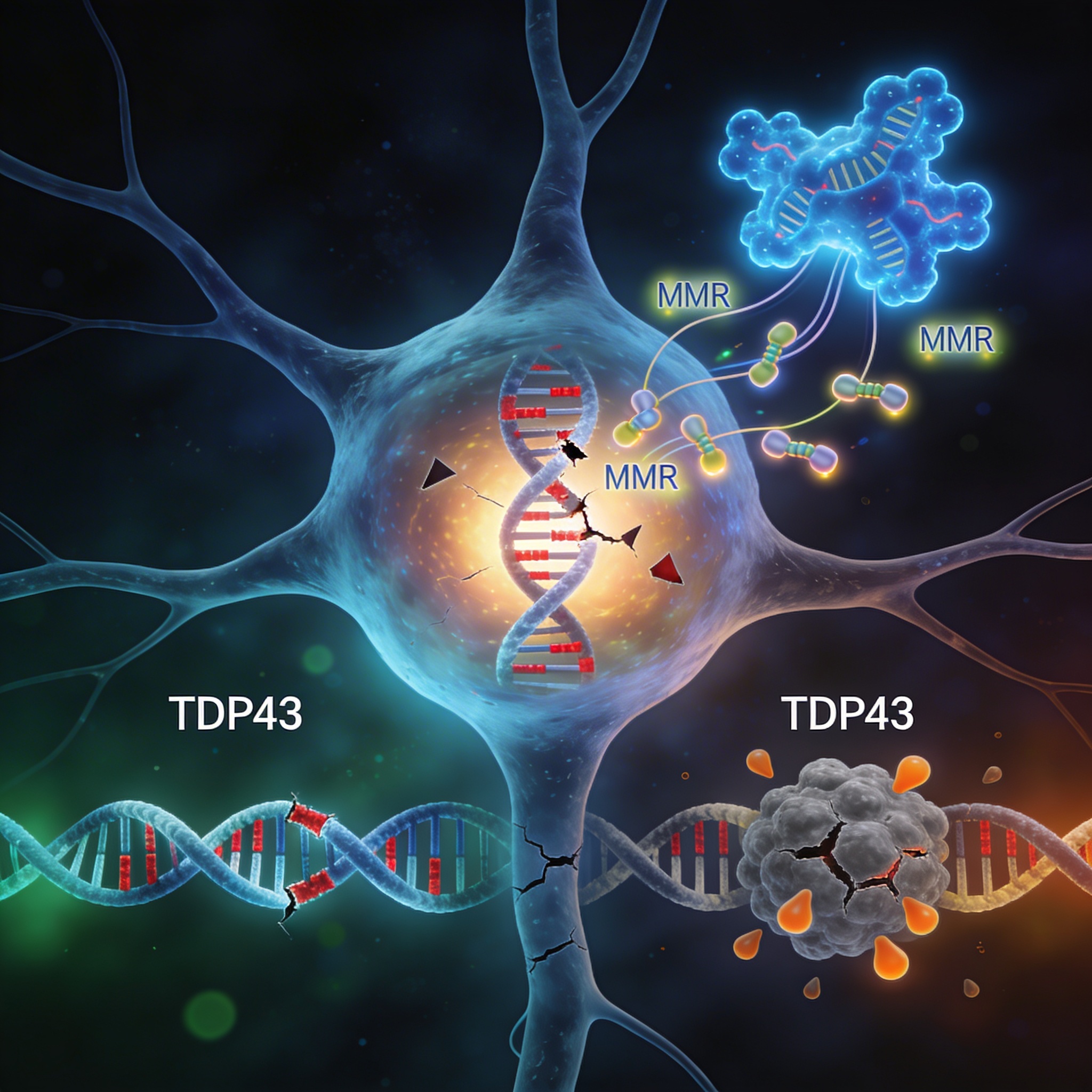
A protein known for its role in neurodegenerative disease has a hidden job: keeping the cell’s DNA repair crew on task. When TDP43 malfunctions—the hallmark of amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD)—that repair machinery grinds to a halt, leaving neurons vulnerable to DNA damage and mutation accumulation.
Key Findings
- TDP43 regulates multiple DNA mismatch repair (MMR) genes: When TDP43 is depleted, expression of MLH1, MSH2, MSH3, MSH6, and PMS2 all drop significantly in both dividing cells and neurons.
- 30% reduction in transcript stability: TDP43 controls the half-life of MMR gene transcripts; when TDP43 is knocked down, mRNA for MLH1 decays 38% faster and MSH6 decays 36% faster in neuronal cells.
- DNA damage accumulates in ALS models: Mice expressing mutant TDP43 show increased markers of unrepaired DNA breaks (γH2AX) in brain tissue, and this damage is reversed when MMR genes are restored.
- Genome-wide analysis maps the network: Across 33 cancer types in the TCGA database, TDP43 expression correlates strongly with MMR gene expression, particularly MSH6 and MSH2, suggesting a conserved regulatory role.
- TDP43 specifically binds MMR transcripts: The protein recognizes intronic sequences in MLH1 and MSH6 pre-mRNA, controlling whether these genes are properly spliced and exported from the nucleus.
- Rescue reverses pathology: Restoring MMR gene expression in TDP43-deficient cells reduces DNA damage accumulation and improves cell survival after oxidative stress.
Source: Nucleic Acids Research (2025) | Provasek et al.
A Double-Duty Protein with Consequences
For decades, TDP43 was primarily known as an RNA-binding protein involved in splicing and gene expression—functions that happen in the nucleus. When it accumulates in the cytoplasm and forms aggregates, neurons die, and the result is ALS or FTD. But a new piece of this puzzle has emerged: TDP43 also acts as a guardian of genomic fidelity, controlling the very genes that catch and correct DNA mistakes before they become mutations.
The discovery shifts how researchers think about these diseases. It’s not just that neurons lose a general RNA-processing protein. They also lose the ability to repair DNA properly—a consequence that may explain why patients with ALS or FTD face elevated cancer risk and why aging neurons are particularly vulnerable to dysfunction.
How TDP43 Controls the Repair Machinery
The study began with a simple question: what happens to DNA repair genes when TDP43 levels drop? Researchers knocked down TDP43 in human neuroblastoma cells and tracked the expression of six core mismatch repair (MMR) proteins—MLH1, MSH2, MSH3, MSH6, PMS2, and TARP (also called P582).
The result was striking. Without adequate TDP43, expression of all five major MMR genes plummeted. MLH1 fell by 50%, MSH3 dropped by 70%, and the others declined proportionally. This wasn’t due to cells dying or dividing at different rates; the reduction was specific to transcript levels.
The mechanism turned out to be more elegant than a simple “off switch.” TDP43 binds directly to the pre-mRNA of MMR genes, controlling how they are spliced—the process by which cells remove non-coding introns and glue exons together to form mature mRNA. When TDP43 is absent or aggregated, this splicing goes awry. Some transcripts fail to leave the nucleus. Others are improperly processed, leading to degradation.
In neuronal cells derived from iPSCs (induced pluripotent stem cells), the effect was even more pronounced. As cells matured from neural progenitor cells to motor neurons, the dependence on TDP43 for MMR gene expression increased. This suggests that mature neurons—the very cells that die in ALS—are exquisitely dependent on TDP43 to keep their repair machinery running.
When Repair Fails, Mutations Pile Up
So what happens when this repair system breaks down? Researchers used ALS mouse models to find out. Mice carrying a mutant TDP43 gene (Tdp43ΔNLS) that mislocalizes to the cytoplasm showed a striking pattern: increased DNA damage markers in the brain, particularly in the spinal cord where motor neurons cluster.
The damage appeared as accumulation of unrepaired double-strand breaks, marked by the protein γH2AX. When researchers restored expression of MMR genes in these damaged cells, the DNA damage decreased and cell survival improved. This demonstrated a causal link: TDP43 dysfunction leads to MMR loss, which leads to DNA damage accumulation.
The implications extend beyond the lab. A comprehensive analysis of cancer genomes revealed that TDP43 expression strongly predicts MMR gene expression across 33 different tumor types. In cancers where TDP43 levels are low, MMR genes are also suppressed—a pattern that could explain why tumors with dysfunctional TDP43 accumulate mutations faster and become more aggressive.
[Insert image: pathway diagram showing TDP43 binding → MMR gene expression → DNA repair capacity; with TDP43 pathology leading to reduced MMR and DNA damage accumulation]
A Shared Vulnerability Between Neurodegeneration and Cancer
This research opens a surprising window into why ALS and FTD patients sometimes develop cancer—or conversely, why cancer patients on chemotherapy sometimes show neurological symptoms. The same cellular machinery that prevents mutations in DNA repair-dependent cancer cells is also essential for keeping neurons alive.
In neurons, unrepaired DNA damage triggers cell death by apoptosis or leads to senescence (permanent cell cycle arrest). In cancer cells, the same unrepaired damage can paradoxically trigger metastasis or treatment resistance. The TDP43-MMR link appears to be a critical pivot point: losing this connection can push a cell toward degeneration (in neurons) or transformation (in tumors).
The study also examined whether TDP43 itself might be mutated or dysregulated in cancer. Analysis of the TCGA database revealed that in some tumors—particularly high-grade gliomas and adenocarcinomas—TDP43 is downregulated, consistent with the hypothesis that tumor cells might “silence” TDP43 to increase mutation rates and fuel adaptability.
Limitations and Future Directions
The study is comprehensive but has important boundaries. Most experiments used cell cultures and mouse models; the mechanisms may differ subtly in human brains. Additionally, TDP43’s role in MMR regulation appears to depend on cell type and developmental stage—mature neurons show stronger dependence than dividing progenitor cells.
A key remaining question is whether restoring MMR gene expression alone would be sufficient to slow neurodegeneration in ALS. The current study shows it reduces DNA damage, but reversing established neuronal loss is a much higher bar. Moreover, TDP43 plays roles beyond MMR regulation—it affects RNA splicing for hundreds of genes—so a therapeutic approach might need to address multiple pathways simultaneously.
Nevertheless, the findings suggest a new target for intervention: stabilizing or restoring MMR gene expression in the context of TDP43 pathology. This could be achieved through drugs that enhance RNA stability, splice-correcting antisense oligonucleotides, or gene therapy approaches tailored to the nervous system.
Why This Matters Now
The research reframes TDP43 from a single-function protein to a critical hub controlling genome stability across multiple cell types. In neurons, loss of TDP43 function means loss of DNA repair capacity, which accelerates aging-like degeneration. In tumors, TDP43 loss may explain increased mutational burden and treatment resistance.
For patients with ALS and FTD, this work offers both a mechanistic explanation for some of the cellular dysfunction and a potential new direction for therapy. It also highlights why these diseases are so difficult to treat: they attack multiple overlapping systems simultaneously. Any successful intervention may need to restore not just TDP43 function broadly, but specifically its ability to regulate the repair machinery that keeps cells—especially long-lived neurons—from accumulating lethal mutations over time.
Citation: Provasek VE, Bacolla A, Rangaswamy S, Kodavati M, Mitra J, Yusuf IO, Malojirao VH, Vasquez V, Britz GW, Li GM, Xu Z, Mitra S, Garruto RM, Tainer JA, Hegde ML. RNA/DNA-binding protein TDP43 regulates DNA mismatch repair genes with implications for genome stability. Nucleic Acids Research. 2025;53:gkaf920. DOI: 10.1093/nar/gkaf920
Authors’ affiliations: Division of DNA Repair Research, Center for Neuroregeneration, Department of Neurosurgery, Houston Methodist Research Institute, Houston, TX; School of Medicine, Texas A&M University; Department of Molecular and Cellular Oncology, University of Texas MD Anderson Cancer Center; Department of Biochemistry and Molecular Biotechnology, University of Massachusetts Chan Medical School; Department of Neurosurgery and Department of Neuroscience, Weill Cornell Medical College.





