TL;DR: A 2026 study in Nature Communications found that in mice, stress from inflammation, forced cell division, cancer-like pressure, and aging activated MLKL in blood-forming stem cells without causing widespread cell death. Instead, active MLKL built up in mitochondria, damaged the cells’ energy machinery, and made the surviving stem cells age faster.
Key Findings
- Several stresses converged on the same pathway: Inflammation, 5-FU replication stress, oncogenic stress, and natural aging all activated the RIPK3-MLKL pathway in hematopoietic stem cells, the blood-forming stem cells that rebuild the immune and blood system.
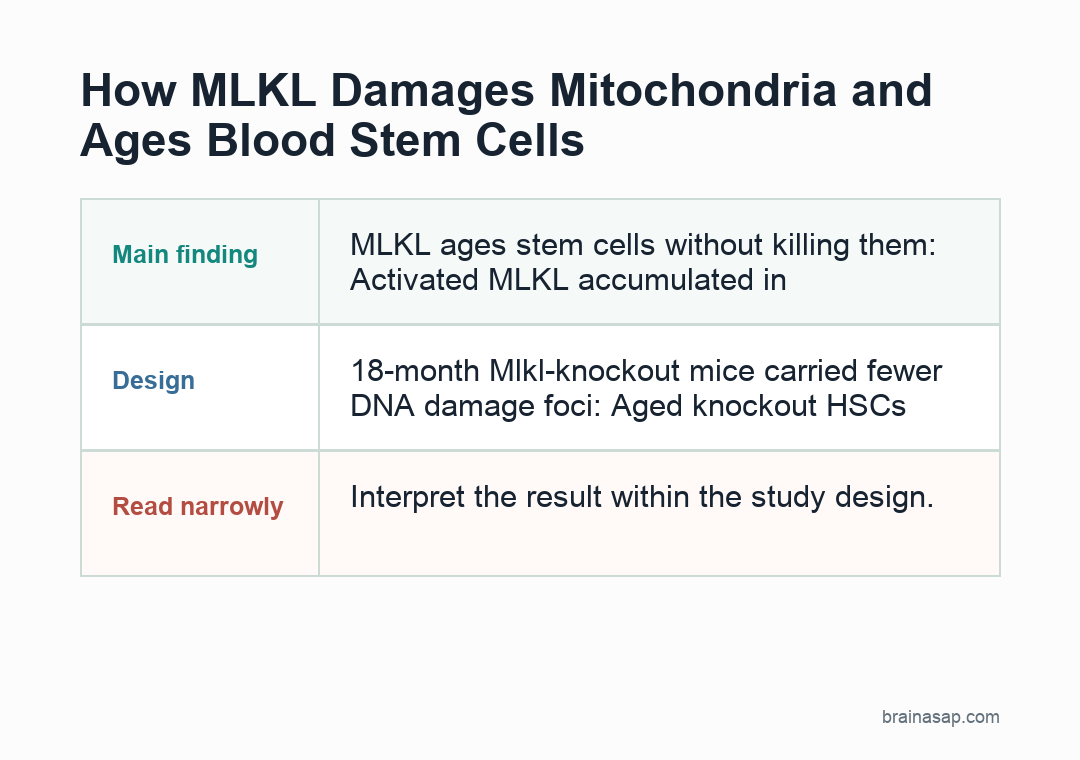
- MLKL acted through mitochondria: MLKL activation increased after inflammatory triggers and 5-FU stress, but stem-cell numbers did not collapse; activated MLKL instead accumulated in mitochondria and reduced energy metabolism.
- 18-month knockout mice aged better: Blood-forming stem cells from 18-month-old Mlkl knockout mice carried fewer γH2AX DNA-damage foci and produced better regenerative and B/T-cell output than aged wild-type stem cells.
- Serial transplants tested stem-cell durability: MLKL-deficient stem cells contributed more strongly to recipient blood systems across primary, secondary, and tertiary transplantation, a hard test of whether stem cells can keep regenerating under repeated stress.
- 17 vs 19 MDS recipients: In a RUNX1S291fs myelodysplastic-syndrome model, recipients of MLKL-deficient donor HSCs survived longer, extending the pathway beyond “normal aging” into stress-linked blood disease risk.
Source: Nature Communications (2026) | Yamada et al.
MLKL is supposed to be an executioner.
In classic necroptosis, it punches holes in membranes and helps finish off a dying cell.
This study found a stranger version of the mechanism: in blood-forming stem cells, MLKL switched on repeatedly without causing cell death, and instead behaved more like an aging accelerator.
Why an Executioner Protein Was Showing Up in Living Stem Cells
Hematopoietic stem cells sit at the top of the blood system. They are the cells that keep making fresh immune cells, red blood cells, and platelets for decades.
Three details anchor the result:
- Several stresses converged on the same pathway: Inflammation, 5-FU replication stress, oncogenic stress, and natural aging all activated the RIPK3-MLKL pathway in hematopoietic stem cells, the blood-forming stem cells that rebuild the immune and blood system
- MLKL activated without mass cell death: MLKL activation increased after inflammatory triggers and after 5-FU, a chemotherapy drug used here to force blood stem cells into regenerative stress, but stem-cell numbers did not collapse
- 18-month knockout mice aged better: Blood-forming stem cells from 18-month-old Mlkl knockout mice carried fewer γH2AX DNA-damage foci and produced better regenerative and B/T-cell output than aged wild-type stem cells
With age and repeated stress, many of these stem cells survive but lose quality.
They regenerate less effectively and produce fewer lymphoid cells, the branch that gives rise to B cells and T cells.
The authors started with a puzzle inside that tradeoff. The RIPK3-MLKL pathway is best known as the machinery of necroptosis, a programmed form of inflammatory cell death.
If that pathway turns on in stem cells, the obvious expectation is that the cells die. But old stem cells do not simply disappear.
They accumulate in a damaged, less capable state. Yamada and other researchers therefore tested whether MLKL can harm stem-cell function without triggering full necroptotic death.
That distinction is central to the result because aging biology often involves cells that remain alive but work poorly.
In this study, activated MLKL did not mainly eliminate blood-forming stem cells; it helped turn surviving stem cells into weaker regenerators.
How Inflammation and 5-FU Turned On MLKL Without Killing Blood Stem Cells
The first striking result is how selective the activation was.
Using a FRET-based biosensor, a tool that reports when a protein changes activity inside living cells, the team saw MLKL activation rise in HSCs and multipotent progenitors after inflammatory triggers like pIC, LPS, and TNF-α.
HSCs are hematopoietic stem cells, the blood-forming cells at the top of the bone-marrow hierarchy.
Multipotent progenitors are one step more committed: they can still make several blood-cell types, but they are less primitive than true stem cells.
The same pattern appeared under replication stress, meaning stress caused by forcing cells to divide and rebuild the blood system.
A single 5-FU exposure activated MLKL in HSCs, and serial 5-FU treatment pushed the blood system toward age-like changes such as myeloid skewing and impaired stem-cell fitness.
When MLKL was removed, much of that decline softened.
What did not happen is just as important. The knockout did not produce a dramatic rescue of stem-cell survival in the simple “fewer dying cells” sense.
HSC counts stayed broadly comparable, and the paper repeatedly shows that the key phenotype is functional decline, not wholesale depletion.
In other words, MLKL was not just deciding whether stem cells lived or died.
It was shaping the quality of the survivors.
The transplantation experiments tested that functional quality directly. Stem-cell biology can look normal in place and still fail under pressure; donor HSCs lacking MLKL held onto function across harder tests:
- Inflammatory stress: MLKL-deficient HSCs retained better output after immune challenge.
- Serial transplantation: MLKL-deficient donor cells kept contributing more strongly to recipient blood systems across repeated regenerative demand.
- Lymphoid output: B- and T-cell potential was less eroded than in aged wild-type HSCs.
What 18-Month-Old Knockout Mice Kept That Wild-Type Stem Cells Lost
By 18 months, wild-type blood-forming stem cells showed familiar signs of blood-system aging: more myeloid bias, more DNA-damage burden, and worse regenerative performance.
Myeloid bias means the stem cells lean toward producing innate immune cells at the expense of lymphoid cells such as B cells and T cells.
In the Mlkl knockout mice, that aging trajectory was visibly blunted.
One clean marker was γH2AX, a readout of accumulated DNA damage. Aged knockout HSCs carried fewer foci than aged wild-type HSCs, consistent with a less beat-up stem-cell state.
That did not mean the animals stayed “young” in every respect. It meant one specific stress pathway contributed measurably to DNA damage and declining blood-stem-cell performance.
The stronger evidence came from transplantation.
Across five experiments, the authors transplanted HSCs from 3-month-old and 18-month-old mice into recipients and tracked how much of the recipient blood system came from the donor stem cells.
Aged wild-type HSCs performed as expected for old stem cells. Aged knockout HSCs did noticeably better, especially in regeneration and in the ability to produce B cells and T cells.
That is a meaningful distinction.
Many aging papers show associations between old age and damaged stem cells.
This one shows that removing a single stress effector preserves function even after the organism has aged, which is much closer to a causal explanation.
The Mechanism Points to Mitochondria, Not Transcription
The mechanistic twist is that the rescue did not seem to come from broad transcriptional rewiring.
The authors looked for age-related differences in the HSC transcriptome and in chromatin accessibility, and MLKL loss changed surprisingly little there.
That negative finding narrows the mechanism.
Instead, the data point toward mitochondrial localization and metabolic damage.
Activated MLKL accumulated in stem-cell mitochondria, the energy-handling structures inside cells, where it was linked to impaired mitochondrial function and reduced glycolytic flux, a measure of how cells process glucose for energy.
That fits with a broader idea in stem-cell aging: the cells are not just turning the wrong genes on and off.
They are losing the energy-handling capacity that lets them stay functionally young.
This is also where the paper departs from the usual necroptosis narrative. MLKL is often described as the terminal membrane disruptor of a dying cell.
Here it behaves more like a chronic saboteur.
The protein is active, but the finding is not cell lysis.
The finding is a stem cell that still exists, yet is less able to renew itself and less able to produce lymphoid descendants.
The difference is concrete.
Age-related disease is often driven by cells that survive in a dysfunctional state.
A pathway that converts acute stress into long-lived mitochondrial damage can help explain gradual tissue decline better than a pathway that simply kills cells outright.
How a Necroptosis Protein Ended Up Looking Like an Aging Pathway
The broader finding is that inflammation, replication stress, oncogenic stress, and natural aging all converged on the same RIPK3-MLKL pathway.
In this model, those stresses produced the same downstream pattern: cumulative mitochondrial injury and declining stem-cell fitness.
The MDS experiment sharpens that point. When the authors introduced the RUNX1S291fs truncation mutant to model myelodysplastic syndrome, recipients of MLKL-deficient donor HSCs survived longer.
That suggests the pathway is not just about “normal” aging in a tidy lab sense. It may sit at the junction where chronic stress, stem-cell exhaustion, and blood-disease susceptibility meet.
There are still limits. This is a mouse HSC paper, not a human trial, and it focuses on the blood system rather than the brain.
But the mechanism is worth tracking beyond this model.
A protein once treated mainly as a death effector acted here as a stress-to-mitochondria link, converting repeated stress into long-term organelle damage without requiring cell death as the main endpoint.
The simplest summary is also the most informative: MLKL can make a stem cell age functionally without killing it.
Citation: DOI: 10.1038/s41467-026-71060-4. Yamada et al. Non-necroptotic MLKL function damages mitochondria and promotes hematopoietic stem cell aging. Nature Communications. 2026;17:2798.
Study Design: Preclinical animal study
Sample/Model: Mouse blood-forming stem-cell aging, inflammatory stress, 5-FU regenerative stress, serial transplantation, and a RUNX1-mutant myelodysplastic-syndrome model.
Key Statistic: MLKL activation rose in blood-forming stem cells after inflammatory triggers and 5-FU stress without a matching collapse in stem-cell numbers, then accumulated in mitochondria and impaired stem-cell function.
Caveat: Single-study evidence; interpret with the source design and sample.