TL;DR: A 2026 mouse and cell study in Signal Transduction and Targeted Therapy linked ATP11B, a phospholipid-flipping transport protein, to hippocampal iron handling, mitochondrial failure, ferroptosis, and age-related cognitive decline.
Key Findings
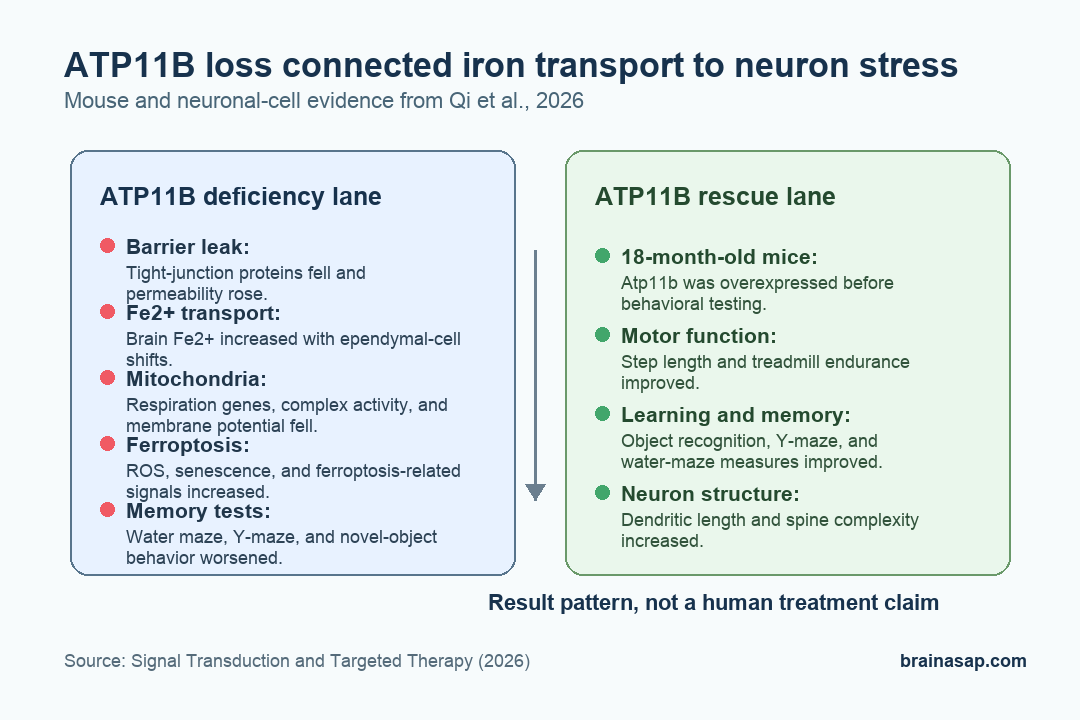
- ATP11B fell with brain aging: Older mice showed lower blood-brain-barrier tight-junction markers and ATP11B emerged from integrated aging, endothelial-cell, and transport-gene analyses.
- ATP11B loss raised brain Fe2+: Atp11b knockout mice had greater blood-brain-barrier permeability, higher brain Fe2+ content, and worse aging-like behavioral signs.
- Memory tests were impaired: Knockout mice spent less time in the Morris water maze target quadrant and performed worse in Y-maze and novel-object memory tasks.
- Mitochondrial respiration weakened: ATP11B-deficient neuronal cells showed lower mitochondrial respiratory-chain gene expression, lower complex I and IV activity, and more fragmented mitochondria.
- ATP11B overexpression improved aged mice: In 18-month-old mice, ATP11B overexpression improved motor behavior, learning and memory tests, dendritic complexity, and ferroptosis-related markers.
Source: Qi et al. (2026)
ATP11B is a phospholipid flippase, meaning it helps move certain lipids between membrane layers. In this paper, researchers treated it as a possible link between aging blood-brain-barrier changes, iron movement, and neuron damage.
Ferroptosis is an iron-related form of cell death driven by lipid damage and oxidative stress. The study tested whether ATP11B loss pushes hippocampal neurons toward that state during brain aging.
ATP11B Emerged From Aging and Transport-Gene Screens
Researchers began with the blood-brain barrier, the vessel barrier that limits what enters brain tissue. Compared with 3-month-old mice, 18-month-old mice had lower expression of Claudin-5, Occludin, and ZO-1 in the hippocampus and cerebral cortex.
Those proteins help maintain tight junctions between barrier cells. Lower levels support the paper’s starting point: aging was associated with weaker barrier integrity in regions relevant to cognition.
The team then combined three lines of evidence:
- Plasma uptake correlations: Genes were compared with blood-to-brain transport behavior.
- Aging endothelial-cell changes: Differentially expressed genes from young and old brain endothelial cells were considered.
- Transport-gene filtering: Transport-related genes were prioritized as possible regulators.
ATP11B ranked as a strong candidate in that integrated screen. The study also reported relatively high ATP11B expression in the choroid plexus, hippocampus, and cerebral cortex, which made it relevant to cerebrospinal-fluid transport and brain-aging biology.
ATP11B Loss Increased Brain Fe2+ and Impaired Memory Tasks
Knockout mice lacking Atp11b showed higher sodium fluorescein permeability, a marker of barrier leakiness. Tight-junction protein levels were lower, and Fe2+ content in brain tissue was higher in a pattern similar to aged mice.
Behavioral testing moved the finding from molecular transport into brain function. Atp11b knockout mice showed signs of accelerated aging, altered social behavior, and weaker memory performance.
- Morris water maze: Knockout mice spent significantly less time in the target quadrant, indicating weaker spatial memory.
- Y-maze: Young and aged knockout mice spent less time in the novel arm and entered it less often.
- Novel-object recognition: Knockout mice, especially aged animals, explored new objects less and returned more often to familiar objects.
The paper interprets those tests as evidence that ATP11B deficiency can produce brain-localized iron accumulation, aging-like decline, and hippocampus-dependent memory impairment. The animal data do not prove the same sequence occurs in humans, but they give the mechanism a behavioral readout.
Ependymal Cells and Neurons Carried the Transport Pattern
Single-cell and single-nucleus sequencing then narrowed the cellular map. Researchers identified seven major brain-cell types and found that Atp11b knockout mice had lower proportions of ependymal cells and neurons.
Ependymal cells line fluid-filled spaces in the brain and help regulate cerebrospinal fluid. In the knockout mice, some ependymal-cell subtypes fell while others rose, suggesting that ATP11B loss changed the balance of cell states involved in fluid and ion handling.
The paper’s transport model is specific: ependymal-cell disruption may alter how Fe2+ moves toward hippocampal neurons. Spatial transcriptomics supported that direction by placing altered genes and cell-state changes into brain tissue context rather than treating the sequencing result as a mixed-cell average.
Mitochondrial Respiration and Quality Control Broke Down
The neuron-level analysis pointed to mitochondria. ATP11B deficiency reduced expression of mitochondrial respiratory-chain genes such as Cox5a and Ndufa4 in excitatory neurons, and ATP11B knockdown reduced Ndufa4 and Ndufb6 expression in SH-SY5Y neuronal cells.
Several mitochondrial readouts moved in the same direction:
- Respiratory complexes: Complex I and complex IV activity were lower after ATP11B knockdown.
- Membrane potential: JC-1 staining showed reduced mitochondrial membrane potential.
- Structure: Mitochondria became more fragmented, with fewer ordered tubular structures.
- Energy metabolism: Basal oxygen consumption, maximal respiration, and ATP-linked respiration were reduced, while lactate increased.
ATP11B loss also shifted mitochondrial quality-control markers. Fission and mitophagy markers rose, and neuronal senescence markers P16, P21, and P53 increased.
Read plainly, the cells were clearing damaged mitochondria while accumulating oxidative-stress markers tied to aging.
KLF4 and YAP Connected Mitochondria to Ferroptosis Genes
The study then tested transcriptional regulation. ATAC-seq and ChIP-seq analyses suggested that ATP11B loss reduced chromatin accessibility at mitochondrial respiration and fusion genes regulated by KLF4, a transcription factor involved in neural development and cellular state control.
At the same time, ATP11B loss changed Hippo pathway activity and increased nuclear movement of YAP1. YAP1 can partner with TEAD transcription factors, and the study found TEAD binding at the transcription start regions of Acsl4, Trp53, and Cdkn1a.
Those genes connect the mitochondrial findings to ferroptosis and cellular senescence. Lactate also entered the model: ATP11B deficiency increased lactate, and researchers linked that metabolite to histone lactylation at ferroptosis and aging-related genes.
ATP11B Overexpression Improved Aged-Mouse Behavior
The strongest intervention test came at the end. Researchers overexpressed Atp11b in 18-month-old mice and compared them with older control animals and 6-month-old young controls.
One month after injection, ATP11B-overexpressing aged mice showed better performance in several tests. Step length increased, treadmill time to exhaustion improved, and spatial-memory measures improved in object-recognition, Y-maze, and Morris water maze tasks.
Brain tissue analysis also moved in the same direction. Dendritic length and spine complexity increased, while hippocampal and cortical assays suggested partial rescue of ferroptosis and mitochondrial-fission effects.
Main limitation: This was a mouse and cell study, not a human treatment trial.
ATP11B overexpression is mechanistic evidence that the pathway can be pushed in a favorable direction in animals. It is not proof that targeting ATP11B will safely slow cognitive aging in people.
Citation: DOI: 10.1038/s41392-026-02652-1. Qi et al. Targeting ATP11B-YAP axis repairs mitochondrial function and inhibits neuronal ferroptosis to attenuate age-related cognitive decline. Signal Transduction and Targeted Therapy. 2026;11:141.
Study Design: Mouse and neuronal-cell mechanistic study using aging comparisons, Atp11b knockout, single-cell and spatial transcriptomics, mitochondrial assays, ferroptosis markers, and overexpression rescue.
Sample Size: Behavioral panels commonly used 10 mice per group; several cell and molecular assays used 3-4 replicates per group.
Key Statistic: Atp11b knockout mice showed significantly weaker Morris water maze, Y-maze, and novel-object recognition performance, while ATP11B overexpression improved aged-mouse motor, memory, and synaptic-complexity measures.
Caveat: The evidence is preclinical, and the paper does not establish whether ATP11B-targeted intervention would be safe or effective for human cognitive aging.