TL;DR: A 2026 Nature Communications study found that chronic tumor necrosis factor alpha (TNF-alpha), an inflammatory cytokine, pushed human hippocampal progenitor cells toward type I interferon activity, lower neuroblast formation, and CXCR3-dependent T-cell recruitment.
Key Findings
- 0.1-1 ng/ml TNF-alpha: The key experiments used chronic low-dose TNF-alpha exposures that the researchers considered biologically relevant for inflammatory microenvironments.
- 22,133 single cells: Single-cell RNA sequencing retained 22,133 high-quality cells across 15 clusters, showing fewer neurogenic clusters after high-dose TNF-alpha exposure.
- Type I interferon loop: TNF-alpha induced IFN-beta and activated IFNAR, the type I interferon receptor, rather than acting only through direct inflammatory pathways.
- CXCR3-dependent T-cell migration: Conditioned media from TNF-alpha-treated hippocampal progenitor cells attracted CD4+ and CD8+ T cells, and a CXCR3 antagonist blunted the migration.
- IFNAR blockade rescued DCX+ cells: Anifrolumab, an IFNAR-blocking antibody, rescued the TNF-alpha-linked loss of DCX+ neuroblasts in the in vitro differentiation model.
Source: Nature Communications (2026) | Nissen et al.
Adult hippocampal neurogenesis is the process by which new neurons form in the hippocampus, a brain region involved in learning, memory, and mood. Inflammation has long been linked to weaker neurogenesis, but the cell-level route from inflammatory cues to fewer new neurons has been harder to pin down.
Nissen and coauthors focused on human hippocampal progenitor cells, the immature cells that can move toward neuronal or glial-like fates. Their question was narrow: could chronic TNF-alpha exposure shift these cells away from neurogenesis and toward an immune-defensive state?
Chronic TNF-alpha Reduced Human Hippocampal Neurogenesis
Researchers used a female-derived human in vitro neurogenesis model, which means the experiments were done in cultured human cells rather than in living people. The cells were exposed to 0.1, 1, or 10 ng/ml TNF-alpha, with the lower two doses used for the main single-cell and rescue experiments.
The model tracked several parts of neurogenesis:
- Hippocampal progenitor cells: The starting cell population used to model early human hippocampal neurogenesis.
- DCX+ neuroblasts: Cells positive for doublecortin (DCX), a marker of immature neuron formation.
- MAP2+ neurons: Cells positive for microtubule-associated protein 2 (MAP2), a marker used for more mature neuronal differentiation.
- Neurite length: A morphology measure that reflects how developing neurons extend early processes.
Chronic TNF-alpha reduced the proportion of DCX+ neuroblasts. At the highest dose, it also reduced MAP2+ cells, and neurite measurements suggested weaker morphological maturation.
Cell-death checks did not support a simple explanation that TNF-alpha merely killed off the neurogenic cells. Instead, the pattern pointed toward a change in cell state and lineage direction.
Single-Cell RNA Sequencing Found a Type I Interferon State
To see what changed inside the cells, researchers ran single-cell RNA sequencing after 48 hours of proliferation exposure and after 7 or 14 days of differentiation. After quality control, the dataset included 22,133 cells grouped into 15 clusters.
High-dose TNF-alpha shifted cells away from neuroblast-like and immature-neuron-like clusters. It also created inflammatory glial/progenitor-like states marked by type I interferon response genes such as ISG15, IFI27, and IFI6.
Type I interferons are immune molecules best known for antiviral defense. Here, the interferon pattern appeared in a sterile inflammation model, meaning no virus was needed to trigger the response.
IFNAR Linked TNF-alpha to CXCL10 and T-Cell Recruitment
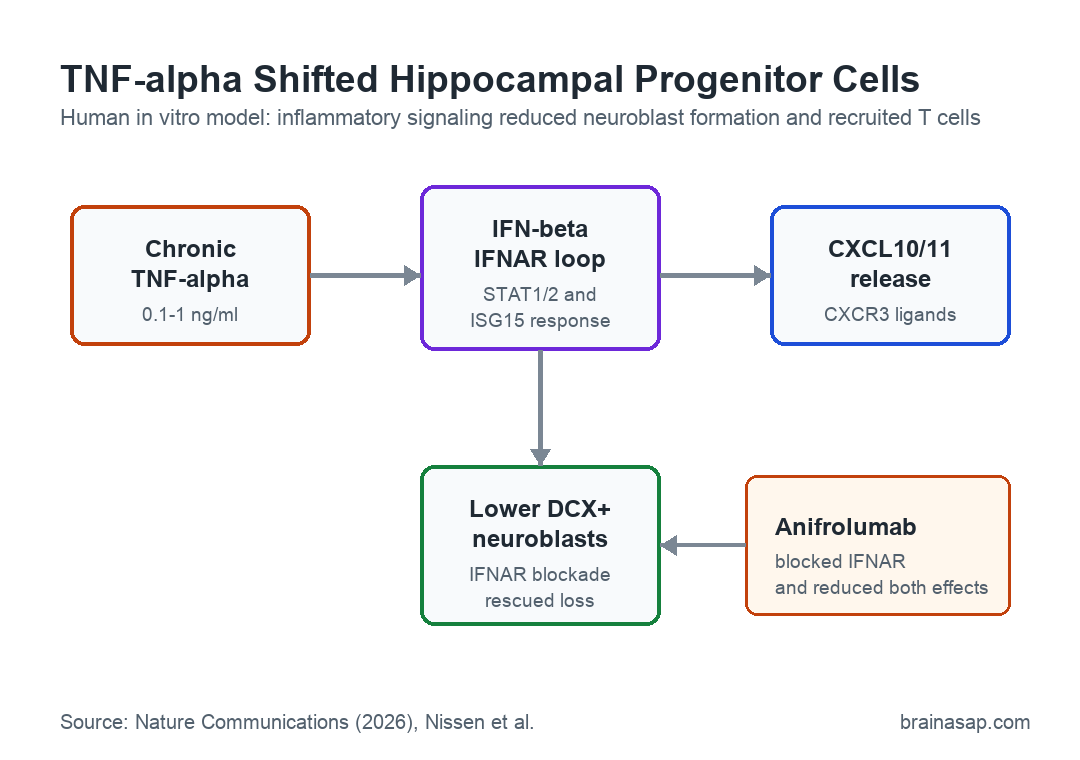
The mechanism centered on an autocrine/paracrine loop. In plain terms, TNF-alpha made the hippocampal progenitor cells produce IFN-beta, and that interferon response then acted back on the same cells or nearby cells through IFNAR, the type I interferon receptor.
Several experiments supported that pathway:
- Delayed STAT activation: STAT1 and STAT2 activation peaked at 3-6 hours, fitting an indirect interferon-mediated mechanism.
- IFNB1 induction: TNF-alpha transiently increased IFNB1, the gene encoding IFN-beta, with a peak around 3 hours.
- IFNAR blockade: Anifrolumab blocked TNF-alpha-induced STAT1+ cell induction and reduced ISG15 upregulation.
- JAK inhibition: Blocking the Janus kinase pathway downstream of IFNAR reduced the interferon-response markers.
The same loop increased chemokine release, especially CXCL10 and CXCL11. These chemokines are ligands for CXCR3, a receptor that helps guide activated T cells toward inflammatory cues.
Conditioned Media Attracted Human CD4+ and CD8+ T Cells
The study moved beyond gene expression by testing T-cell movement. Researchers collected conditioned media from TNF-alpha-treated progenitor cells and from cells differentiated under chronic TNF-alpha exposure.
That conditioned media increased migration of activated primary human CD4+ and CD8+ T cells from three healthy donors. Pretreating the T cells with a CXCR3 antagonist reduced the response, which tied the migration to the CXCL10/CXCL11-CXCR3 route.
IFNAR and JAK blockade also reduced T-cell chemotaxis toward TNF-alpha-conditioned media. This placed type I interferon activity upstream of the chemokine response rather than making CXCL10 release a separate TNF-alpha effect.
Anifrolumab Rescued DCX+ Neuroblast Loss in the Model
The strongest functional rescue came from the IFNAR-blocking antibody anifrolumab. In differentiating hippocampal progenitor cells exposed to 1 ng/ml TNF-alpha, IFNAR blockade fully rescued the loss of DCX+ neuroblasts.
That result does not make anifrolumab a proven neurogenesis treatment. It identifies IFNAR activity as a candidate checkpoint inside this cell model, where one pathway affected both neuroblast formation and inflammatory T-cell recruitment.
The main limits are important:
- In vitro model: The work used cultured human hippocampal progenitor cells, not a human clinical trial or an in vivo brain model.
- Focal dosing uncertainty: Human hippocampal TNF-alpha levels during disease are hard to measure directly, so culture doses may not map neatly onto living tissue.
- Single-cell replication limits: Some single-cell cluster proportions came from one 10x library per condition and timepoint, so the transcriptomic map is mechanistic rather than population-level evidence.
The study still gives a concrete route from chronic inflammation to weaker hippocampal neurogenesis: TNF-alpha can engage IFN-beta and IFNAR activity in human progenitor cells, raise CXCR3 ligands, attract T cells, and reduce DCX+ neuroblast formation.
Citation: DOI: 10.1038/s41467-026-74104-x. Nissen et al. TNF-alpha induces type I IFN signalling to suppress neurogenesis and recruit T cells. Nature Communications. 2026;17:5287.
Study Design: Human in vitro hippocampal neurogenesis model with TNF-alpha exposure, single-cell RNA sequencing, IFNAR/JAK blockade, and T-cell chemotaxis assays.
Sample/Model: Female-derived human hippocampal progenitor cells plus activated primary human T cells from three healthy donors for migration assays.
Key Statistic: Single-cell RNA sequencing retained 22,133 high-quality cells across 15 clusters and identified TNF-alpha-linked interferon-responsive progenitor/glial states.
Caveat: The findings are mechanistic cell-culture evidence and need in vivo validation before clinical treatment claims.