TL;DR: A 2026 study in Nature Metabolism identified brain hyperglycosylation as a metabolic driver of Alzheimer disease in human tissue and mouse models, while glucosamine exposure was linked to worse outcomes in Alzheimer-related dementia records.
Key Findings
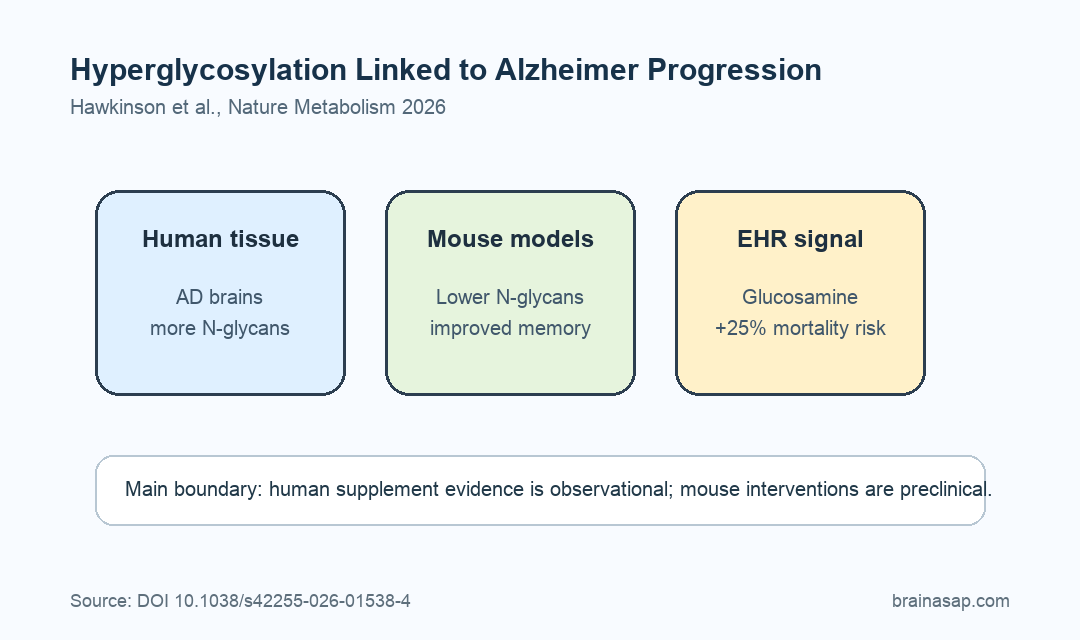
- Human AD brains showed excess glycans: Spatial glycomics found elevated N-glycan abundance in Alzheimer frontal cortex samples compared with matched controls.
- Mouse models replicated the pattern: Hyperglycosylation appeared in 5xFAD and PS19 Alzheimer models, especially in cortex, hippocampus, and thalamus.
- Biosynthesis drove the change: Isotope tracing showed increased glycan biosynthesis rather than impaired degradation or recycling as the main driver.
- Reducing glycosylation helped mice: PGM3 knockdown and NGI-1 treatment lowered brain N-glycans and improved social memory measures in Alzheimer mouse models.
- Glucosamine tracked worse outcomes: In University of Florida Health records, glucosamine use was associated with a 25% higher mortality risk among ADRD patients and higher MCI-to-ADRD conversion.
Source: Nature Metabolism (2026) | Hawkinson et al.
Hyperglycosylation means excess addition of sugar-based glycan structures to molecules such as proteins and lipids.
Hawkinson et al. tested whether that metabolic pattern contributes to Alzheimer disease rather than only appearing after neurodegeneration has begun.
The study combined spatial multiomics in human brain tissue, isotope tracing in mouse models, genetic and drug interventions, dietary glucosamine experiments, and electronic health record analysis.
Spatial Glycomics Found Excess Glycans in Alzheimer Brain Tissue
Researchers first optimized a spatial metabolomics, lipidomics, and glycomics workflow for human frontal cortex. The early technical problem was that human brain lipids interfered with glycan detection, so the team used an extended xylene wash to improve glycan recovery.
After optimization, the analysis compared human Alzheimer brain samples with matched control tissue. Several N-glycan species were markedly elevated across grey and white matter regions in Alzheimer samples.
The human tissue cohort was small for this first spatial comparison, with n = 3 Alzheimer and n = 3 control samples. The strength of the finding came from pairing that human tissue pattern with animal models and mechanistic tests.
The same glycan excess appeared across disease stages and in mouse models:
- Human tissue: Alzheimer samples showed higher representative N-glycan abundance than controls.
- 5xFAD mice: Hyperglycosylation was prominent in Alzheimer-vulnerable brain regions.
- PS19 mice: A tauopathy model showed a related glycosylation phenotype.
Increased Glycan Biosynthesis Drove the Alzheimer Metabolic Pattern
Hyperglycosylation can arise from more glycan production, less glycan degradation, or altered recycling. The team used 13C-glucose isotope tracing to separate those possibilities in 5xFAD mice.
During the pulse phase, 5xFAD brains showed increased enrichment of labeled glycan species compared with wild-type controls. After the chase phase, the pattern did not support impaired degradation as the main explanation.
The interpretation was that increased glycan biosynthesis was the main driver. Pooled LC-MS tracing also showed higher labeled UDP-hexose and UDP-HexNAc, two precursor pools in the N-glycan biosynthetic pathway.
Gene-expression assays supported the same direction. Key glycan biosynthesis genes, including Mgat, Man1a2, and B4galt1, were upregulated in human Alzheimer brains and 5xFAD mouse models.
Reducing N-Glycosylation Improved Social Memory in Alzheimer Mice
The strongest causal tests reduced glycosylation. Researchers targeted PGM3, a hexosamine biosynthetic pathway enzyme, with lentiviral shRNA in 8-month-old female 5xFAD and PS19 mice.
After 2 weeks, brain N-glycan levels decreased and social memory performance improved compared with scrambled-control treatment. The social memory test measured whether mice reduced interaction with a familiar conspecific across repeated exposures.
A second intervention used NGI-1, an oligosaccharyltransferase inhibitor that blocks transfer of N-glycans to nascent proteins. A single intracerebroventricular dose in 8-month-old 5xFAD mice also reduced N-glycan abundance and improved social memory measures.
The intervention findings separate two claims:
- Pathology claim: Excess N-glycosylation was linked to Alzheimer-like brain dysfunction.
- Target claim: Reducing N-glycan biosynthesis improved behavior in mouse models.
- Translation limit: The tested interventions were brain-delivered experimental tools, not approved Alzheimer treatments.
Glucosamine Worsened Alzheimer-Model and EHR Outcomes
Glucosamine is an over-the-counter joint supplement and a precursor feeding glycan-related metabolism. The study tested whether adding glucosamine worsened the disease-linked glycosylation state.
In 5xFAD mice, 2 weeks of oral glucosamine increased brain N-glycan abundance and impaired social memory compared with vehicle-treated 5xFAD controls. The same supplementation did not produce hyperglycosylation or social memory impairment in wild-type mice.
The human EHR analysis used University of Florida Health records from 2012 to 2024. Researchers identified patients with mild cognitive impairment, Alzheimer disease, and Alzheimer-related dementias, then classified glucosamine exposure from physician notes and medication records.
After matching and adjustment, glucosamine use was associated with a 25% increase in mortality risk among ADRD patients. Glucosamine use was also linked to a 25% increase in the proportion of MCI patients transitioning to ADRD.
The Supplement Finding Needs Careful Interpretation
The EHR result is not a randomized trial. People who take glucosamine may differ from non-users in ways that records cannot fully capture, even after matching and adjustment.
The EHR association deserves attention because it aligns with the biology. Human tissue showed hyperglycosylation, mouse models showed increased glycan biosynthesis, reducing glycosylation improved mouse behavior, and glucosamine worsened Alzheimer-model outcomes.
For people with Alzheimer disease or cognitive impairment, the practical step is not self-directed supplement changes based on one study. The appropriate step is to discuss glucosamine use with a clinician, especially when dementia is already diagnosed or progressing.
Citation: DOI: 10.1038/s42255-026-01538-4. Hawkinson et al. Hyperglycosylation is a metabolic driver of Alzheimer’s disease. Nature Metabolism. 2026.
Study Design: Multi-method Alzheimer disease metabolism study combining human brain spatial multiomics, mouse isotope tracing, genetic and drug perturbation, glucosamine supplementation, and retrospective EHR analysis.
Sample/Model: Human Alzheimer and control frontal cortex samples, 5xFAD and PS19 mouse models, and University of Florida Health EHR cohorts with MCI and Alzheimer-related dementia diagnoses.
Key Statistic: Glucosamine use was associated with a 25% higher mortality risk among ADRD patients and a 25% higher proportion of MCI-to-ADRD transition in the EHR analysis.
Caveat: The human supplement association is observational, and the causal intervention evidence comes from mouse models and experimental glycosylation tools.