TL;DR: A 2026 study in Nature Communications found that proteotoxic stress can push neurons into karyoptosis, a cell-death pathway marked by nuclear lamina breakdown, nuclear-material expulsion, and dementia-linked tissue signatures.
Key Findings
- Karyoptosis was induced by protein-clearance stress: Blocking autophagic lysosomal clearance caused LaminB1 disruption, nuclear-shape loss, cytoplasmic nuclear material, and later cell death.
- p38-LaminB1 signaling controlled the pathway: The stress kinase p38 MAPK phosphorylated LaminB1, while p38 inhibition reduced nuclear lamina damage in the experimental models.
- ALS/FTD models showed the same pattern: Toxic C9ORF72 dipeptide-repeat proteins triggered karyoptosis-like nuclear changes in neurons and fly models.
- Human dementia tissue contained matching signatures: Post-mortem frontal cortex from frontotemporal lobar degeneration and Alzheimer’s disease cases had more cells with karyoptosis-like features than controls.
- Therapeutic targeting remains early: The work points to p38 MAPK and LaminB1 stability as possible neuroprotection targets, but the evidence is still preclinical and tissue-based.
Source: Casterton et al. reported the study in Nature Communications.
Neurons often die in diseases where damaged or misfolded proteins build up, but ordinary apoptosis does not explain every dying cell seen in aging brains and neurodegenerative disease.
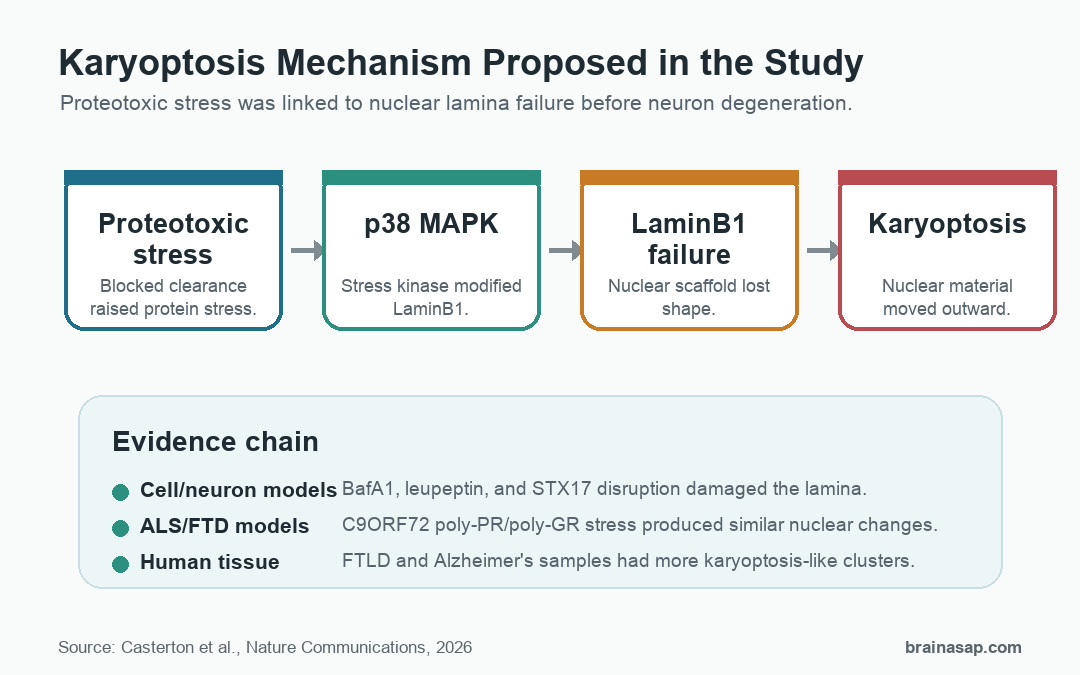
This paper names and tests a different route: karyoptosis, a form of degeneration in which the cell nucleus loses structural support before the cell dies.
Proteotoxic Stress Damaged the Nuclear Lamina Before DNA Damage Rose
The study began with a simple pressure test. Researchers impaired the autophagy-lysosome system, the cellular clearance pathway that helps remove damaged proteins and organelles.
In neuroblastoma cells and rat cortical neurons, that stress produced a sequence of nuclear changes. LaminB1, a structural protein in the nuclear lamina, moved into abnormal cytoplasmic puncta.
Nuclear shape became less regular, and the cell body shrank.
The timing mattered. LaminB1 disruption appeared before stronger markers of DNA damage, which suggests DNA damage was downstream of nuclear lamina failure rather than the first event.
- Stress trigger: BafA1 or related clearance disruption increased proteotoxic load.
- Nuclear response: LaminB1 left the normal nuclear rim pattern and appeared in cytoplasmic puncta.
- Cell consequence: Nuclear shape deteriorated, cells atrophied, and later death markers increased.
The researchers also found nuclear material outside the cell in large extracellular vesicles. Those vesicles carried DNA and nuclear proteins, including LaminB1, which fit the idea that karyoptosis involves expulsion of nuclear material rather than a clean apoptotic shutdown.
p38 MAPK and LaminB1 Formed the Main Control Point
The pathway narrowed around p38 MAPK, a stress-activated kinase. In the cell models, p38 inhibition reduced LaminB1 nuclear-shape abnormalities and reduced cytoplasmic LaminB1 puncta.
Biochemical experiments supported a direct link: p38 phosphorylated a LaminB1 fragment, and the paper tied that phosphorylation to LaminB1 stability. A non-phosphorylatable LaminB1 mutation resisted loss of LaminB1 in fibroblasts, and a corresponding LaminB mutation in flies helped test the same logic in a whole organism.
The fly work is important because it moved the pathway beyond dish-only biology. A LaminB mutation designed to resist the equivalent p38 phosphorylation site raised LaminB levels during aging and improved survival when autophagic clearance was genetically impaired.
- Autophagy failure: Protein-clearance stress pushed the nucleus into a vulnerable state.
- p38 activation: The stress kinase modified LaminB1.
- Lamina instability: The nuclear scaffold became less stable.
- Karyoptosis: Nuclear degeneration and material expulsion preceded cell loss.
ALS/FTD Models Connected Karyoptosis to Protein-Aggregation Disease
The study then asked whether the same nuclear pathway appears under disease-relevant protein stress. The researchers used C9ORF72 dipeptide-repeat proteins, especially poly-PR, which are tied to amyotrophic lateral sclerosis and frontotemporal dementia biology.
In primary cortical neurons, poly-PR caused LaminB1 puncta, abnormal nuclear shape, and TUNEL-positive DNA damage without clear caspase-3 activation. That combination fits the paper’s central claim: the cells showed a death pattern that was not explained by ordinary apoptosis alone.
Drosophila models gave a second test. Flies expressing toxic poly-PR or poly-GR showed LaminB nuclear-shape changes and proteotoxic stress.
Reducing p38 activity partially rescued motor impairment and lifespan in those models. The LaminB phosphorylation-site mutation also improved nuclear shape and survival in the poly-PR setting.
Human iPSC-derived neurons added another relevant check. BafA1 stress reduced cell number and lowered the nuclear-to-cytoplasmic ratio of TDP-43, a pathology-linked protein in ALS/FTD. p38 inhibition significantly rescued both measures.
Dementia Brain Tissue Contained More Karyoptosis-Like Cells
The strongest disease-context result came from post-mortem human frontal cortex. The researchers stained tissue from people with frontotemporal lobar degeneration, people with Alzheimer’s disease, and age- and sex-matched controls.
Instead of manually choosing cells, they used single-cell morphology and LaminB1 measurements with unsupervised clustering. The algorithm was blind to whether a cell came from control, FTLD, or Alzheimer’s tissue.
Cells with karyoptosis-like features were more common in the disease groups. In one analysis, early and late karyoptosis clusters accounted for 35-37% of neurons in FTLD or Alzheimer’s tissue compared with 17% in controls.
The added disease-associated effect was roughly 18-20% cell degeneration in those dementia groups.
A separate dataset found an extra 8-12% late-stage karyoptotic cell degeneration in FTLD and Alzheimer’s disease. These numbers do not prove karyoptosis is the only cause of neuronal loss, but they make it hard to treat the pathway as a cell-culture artifact.
Nuclear Lamina Stability Became a Testable Target
Karyoptosis gives researchers a more specific way to talk about nuclear failure during neurodegeneration. The pathway links proteotoxic stress, autophagy failure, p38 MAPK, LaminB1, and dementia-tissue morphology.
That does not make p38 inhibition a ready treatment. p38 MAPK has broad roles in stress signaling and inflammation, and the paper does not test a human therapy.
The immediate value is mechanistic: nuclear lamina stability becomes a measurable part of neuronal vulnerability.
- What the paper supports: Karyoptosis is a defined nuclear-degeneration pathway that can follow proteotoxic stress.
- What remains open: How much this pathway drives clinical symptoms across different diseases and brain regions is still unresolved.
- What comes next: Better tissue measurement and pathway-specific intervention studies are needed before therapeutic claims are strong.
Some neurons in dementia may not simply be dying through the usual apoptosis story. They may be losing nuclear structure through a stress pathway that can now be measured, modeled, and tested more directly.
Citation: DOI: 10.1038/s41467-026-73802-w. Casterton et al. Karyoptosis mediates cell death and neurodegeneration upon proteotoxic stress. Nature Communications. 2026;17:5135.
Study Design: Cell, primary-neuron, Drosophila, human iPSC-derived neuron, and post-mortem human brain tissue experiments.
Sample/Model: SH-SY5Y cells, rat cortical neurons, C9ORF72 dipeptide-repeat fly models, engineered human neurons, and post-mortem frontal cortex from FTLD, Alzheimer’s disease, and control cases.
Key Statistic: Karyoptosis-like clusters represented 35-37% of neurons in FTLD or Alzheimer’s tissue versus 17% in controls in one tissue analysis.
Caveat: The therapeutic evidence is preclinical; the human data show tissue association, not proof that blocking karyoptosis improves disease outcomes.