TL;DR: A 2026 systematic review and meta-analysis in Neuropsychopharmacology found that dopamine D2-like receptor activation suppressed glucose-stimulated insulin secretion (GSIS), insulin release after high glucose, in animal pancreatic cells, suggesting one peripheral route by which antipsychotic dopamine blockade can disturb glucose control beyond weight gain.
Key Findings
- 39 eligible studies: Researchers screened 12,457 citations and included 39 preclinical in vitro or ex vivo studies, with 37 included in meta-analysis.
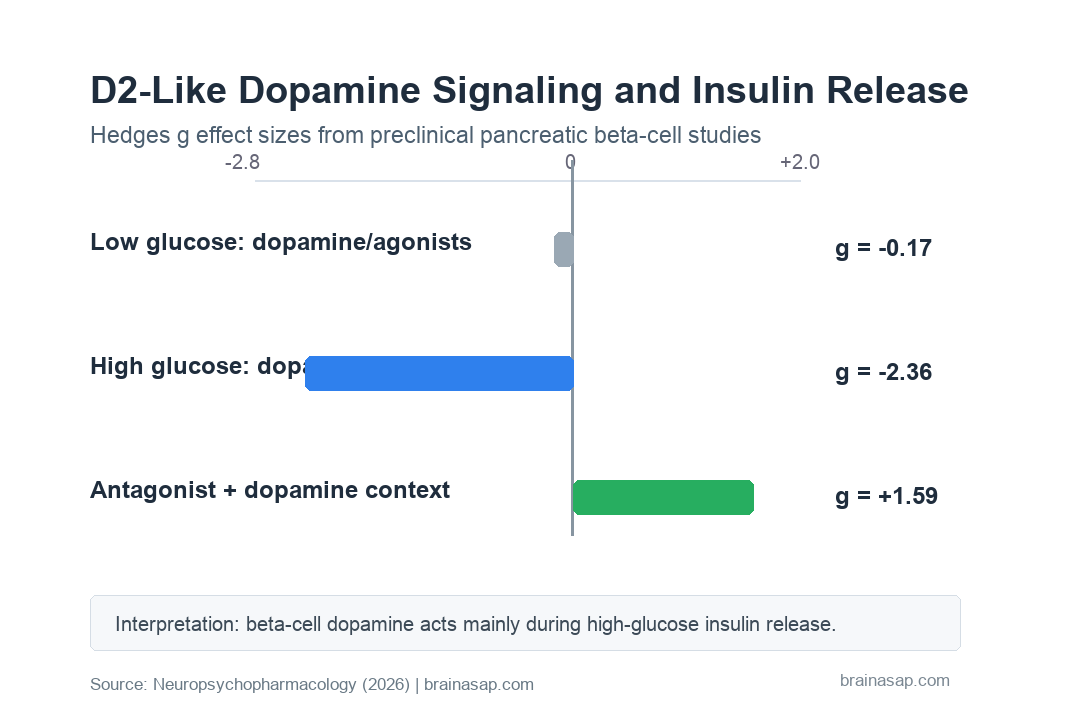
- No low-glucose effect: Dopamine or D2-like agonists did not significantly change basal insulin secretion under low-glucose conditions (Hedges’ g = -0.17; p = 0.36).
- High-glucose insulin suppression: Under high glucose, dopamine and D2-like agonists robustly reduced insulin secretion in rodent studies (g = -2.36; 95% CI, -2.77 to -1.96).
- Rabbit studies agreed: 5 rabbit studies also showed dopamine or L-DOPA suppressed GSIS (g = -1.98; 95% CI, -2.88 to -1.09).
- Antagonists alone were neutral: D2-like receptor antagonists by themselves did not significantly alter insulin secretion (g = -0.25; p = 0.25).
- Antagonists blocked dopamine’s brake: When dopamine or D2-like agonists were present, antagonists increased insulin secretion relative to agonist treatment alone (g = 1.59; p = 0.0002).
Source: Neuropsychopharmacology (2026) | Al-Hasani et al.
Antipsychotic Metabolic Risk May Involve Pancreatic Dopamine Receptors
Dopamine D2-like receptors are usually discussed in the brain because many antipsychotic drugs work by blocking them. This review asks a more peripheral question: what happens when similar dopamine signaling is considered in pancreatic beta cells, the insulin-producing cells that respond to blood glucose?
The clinical background is clear. People prescribed antipsychotic medications have higher risk of dysglycemia, impaired glucose control that can include insulin resistance, impaired glucose tolerance, and type 2 diabetes.
Weight gain explains part of that risk, but not all of it. The review focuses on a possible weight-independent mechanism: dopamine signaling inside the pancreas acts as a glucose-dependent brake on insulin release, and dopamine-blocking drugs can interfere with that brake.
39 Preclinical Studies Tested Dopamine and Insulin Secretion
The review followed PRISMA methods and was registered in PROSPERO. Researchers searched PubMed, Embase, and PsycINFO through September 22, 2025.
Eligible studies had to test dopamine, D2-like receptor agonists, or D2-like receptor antagonists in isolated pancreatic islets or beta-cell lines. The studies also needed quantitative insulin-secretion data under low-glucose and high-glucose conditions.
Researchers excluded whole-animal studies because systemic factors could obscure beta-cell-specific effects. They also excluded dopamine D1-like receptor studies and antagonist experiments where the drugs had substantial activity at other receptor systems known to affect insulin secretion.
- Screening scale: The team screened 12,457 citations before identifying 39 eligible studies.
- Meta-analysis set: 37 studies provided data suitable for meta-analysis.
- Species base: Most studies used rodent beta-cell lines or pancreatic islets, with 5 rabbit studies and a smaller human-islet evidence base reviewed narratively.
- Main compounds: Dopamine, quinpirole, bromocriptine, and L-DOPA were grouped as dopamine or D2-like agonist exposures; antagonists included haloperidol, sulpiride, and raclopride.
This makes the paper a mechanistic synthesis rather than a patient-outcome study. It does not prove that a specific antipsychotic causes diabetes through beta cells, but it quantifies a receptor-level pathway that could contribute.
Dopamine Suppressed GSIS Only When Glucose Was High
The most important distinction was glucose state. Under basal, low-glucose conditions of 2.5-5.6 mmol/L, dopamine or D2-like agonists did not significantly change insulin secretion.
The pooled low-glucose effect was Hedges’ g = -0.17, with a 95% confidence interval from -0.54 to 0.19 and p = 0.36. Heterogeneity was negligible, suggesting the low-glucose null result was fairly consistent across included rodent experiments.
High glucose produced a different pattern. Across 84 experiments from 29 rodent studies, dopamine and D2-like agonists significantly reduced GSIS.
The pooled high-glucose effect was large: g = -2.36, with a 95% confidence interval from -2.77 to -1.96 and p < 0.0001. Even after trim-and-fill adjustment for possible publication bias, the inhibitory effect remained significant.
Meta-regression supported the biological logic. Higher glucose concentration and higher dopamine dose were both associated with stronger insulin suppression during GSIS.
D2-Like Antagonists Reversed Dopamine’s Insulin Brake
D2-like receptor antagonists did not significantly change insulin secretion when given alone. The pooled estimate was g = -0.25, with a 95% confidence interval from -0.68 to 0.18 and p = 0.25.
Mechanistically, the antagonists were not simply pushing insulin secretion up or down in isolation. Their effect depended on whether dopamine or a D2-like agonist was also present.
When antagonists were co-administered with dopamine or selective D2-like agonists, they attenuated GSIS inhibition. In practical terms, they lifted part of dopamine’s brake on insulin release.
- Co-administration result: 15 comparisons from 10 rodent studies found increased insulin secretion compared with agonist treatment alone.
- Pooled effect: The primary estimate was g = 1.59, with a 95% confidence interval from 0.76 to 2.42 and p = 0.0002.
- Glucose moderator: Each 1 mmol/L increase in glucose was associated with a 0.21-unit increase in Hedges’ g for antagonist effects.
- Publication-bias caution: Trim-and-fill adjustment weakened the antagonist-plus-agonist estimate, so this part of the evidence should be interpreted carefully.
Human-islet studies were not numerous enough for meta-analysis, but the narrative pattern was broadly consistent. Dopamine and D2-like agonists generally suppressed GSIS, while D2 antagonists increased insulin release in high-glucose human-islet conditions.
Beta-Cell Dopamine Signaling Could Explain Weight-Independent Dysglycemia
Pancreatic beta cells can synthesize dopamine from L-DOPA. In the proposed mechanism, locally produced dopamine acts through D2 and D3 receptors to fine-tune insulin output when glucose is high.
That brake can protect beta cells from overstimulation after glucose exposure. D2-like receptor activation couples to G alpha i/o signaling, lowers cyclic AMP, reduces calcium influx, and suppresses insulin granule release.
Antipsychotic drugs that strongly block D2-like receptors can disrupt that local feedback. If sustained over time, the review argues, the result can contribute to hyperinsulinemia, insulin resistance, impaired glucose tolerance, and type 2 diabetes risk.
This is not the only metabolic route for antipsychotic side effects. Appetite, weight gain, hypothalamic signaling, histamine and muscarinic receptors, inflammation, sleep, activity, and illness-related factors can also matter.
The beta-cell mechanism adds a second point: metabolic monitoring should not wait until weight has already changed. Glucose and lipid surveillance may matter even when body weight is stable.
Antipsychotic Selection and Monitoring Need More Than Weight
The review’s clinical implication is not that dopamine blockade should be avoided in all patients. Antipsychotics can be essential treatments for psychosis, bipolar disorder, severe depression with psychotic features, and other high-risk conditions.
The more useful clinical point is risk management. If peripheral D2-like blockade contributes to glucose dysregulation, then body weight alone is an incomplete safety marker.
- Baseline measurement: Fasting glucose, HbA1c, lipids, weight, waist measures, and blood pressure can establish a safer starting point before treatment.
- Early follow-up: Glucose markers deserve checking even before major weight gain appears, especially in patients with family history, prediabetes, or prior antipsychotic metabolic effects.
- Drug choice: Medication selection can weigh psychiatric efficacy, receptor profile, prior response, and metabolic vulnerability rather than treating metabolic risk as a late side issue.
- Future treatments: Non-dopaminergic antipsychotic strategies, such as muscarinic approaches, become more important if they preserve efficacy while lowering metabolic burden.
The review also points to bromocriptine, a D2-like agonist used in type 2 diabetes treatment, as biological context for the insulin-suppression pathway. That comparison supports the receptor logic while keeping clinical treatment decisions separate from this preclinical mechanism.
Preclinical Evidence Limits the Antipsychotic Insulin Claim
The paper’s strongest evidence comes from controlled preclinical models, not from clinical diabetes outcomes. That gives the review mechanistic precision but limits direct translation.
Several constraints are important:
- Mostly rodent data: The main meta-analyses relied heavily on rodent beta-cell and pancreatic-islet studies.
- Limited human islets: Only 4 human-islet studies were identified, and they could not be pooled quantitatively.
- High heterogeneity: Glucose concentrations, compound types, doses, and experimental systems varied across studies.
- Physiologic-dose concern: Many in vitro studies used micromolar dopamine concentrations that may exceed normal intra-islet levels.
- Other receptors remain relevant: Adrenergic receptors, alpha cells, central dopamine pathways, histamine receptors, and muscarinic receptors may also contribute to antipsychotic metabolic effects.
These limits keep the claim mechanistic rather than diagnostic. The review identifies a plausible beta-cell dopamine pathway, but patient-level studies are needed to test whether that pathway predicts glucose changes after starting or switching antipsychotic medications.
A strong next study would pair clinical metabolic monitoring with receptor-informed drug exposure, pancreatic or endocrine biomarkers, and enough follow-up to separate acute insulin changes from later weight-mediated effects.
Citation: DOI: 10.1038/s41386-026-02388-0. Al-Hasani et al. Dopaminergic modulation of pancreatic beta-cell insulin secretion and implications for antipsychotic-induced glucose dysregulation: a systematic review and meta-analysis. Neuropsychopharmacology. 2026.
Study Design: Systematic review and random-effects meta-analysis of preclinical in vitro and ex vivo pancreatic islet and beta-cell studies.
Sample/Model: 39 eligible studies; 37 included in meta-analysis, mostly rodent and rabbit pancreatic beta-cell or islet experiments.
Key Statistic: Dopamine and D2-like agonists reduced high-glucose insulin secretion in rodent studies (Hedges’ g = -2.36; 95% CI, -2.77 to -1.96).
Caveat: Mostly preclinical evidence with limited human-islet data, high heterogeneity, possible publication bias, and no direct patient outcome test.