How Lecanemab Clears Brain Amyloid: The Microglia Key
TL;DR: Lecanemab, the first Alzheimer’s antibody to slow cognitive decline, works by activating immune cells called microglia through a specific immune signaling pathway, with the molecule SPP1/osteopontin playing a critical role in triggering the brain’s own cleanup machinery.
When lecanemab was approved by the FDA, it sparked hope but also raised a fundamental question: how exactly does binding to amyloid plaques translate into their removal? Researchers had assumed direct antibody-mediated clearance, but the real mechanism turned out far more elegant—and dependent on the brain’s own immune guardians.
Key Findings
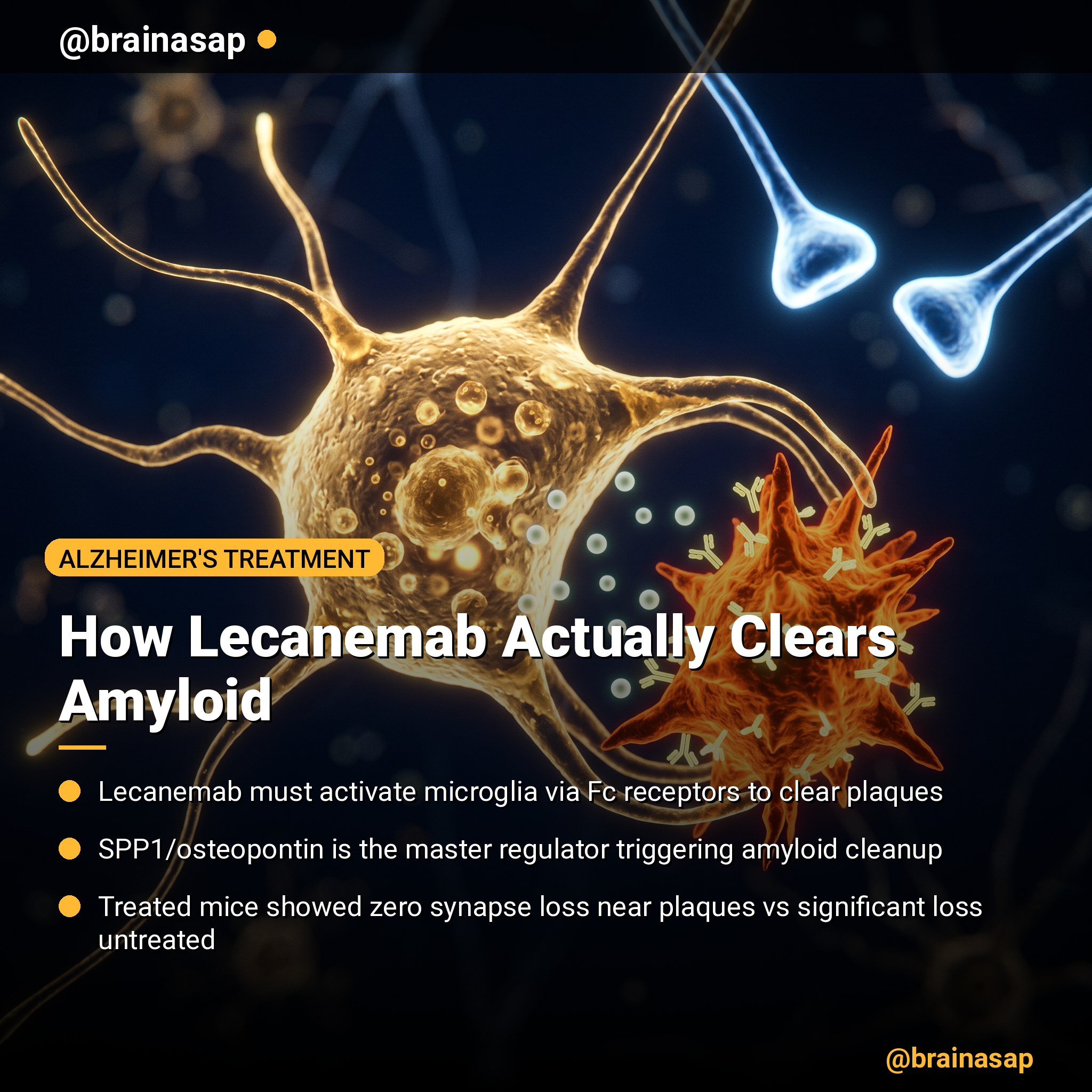
- Fc fragment essential for clearance: Lecanemab’s therapeutic effect requires intact Fc regions that activate microglial receptors; antibodies without functional Fc fragments failed to reduce amyloid plaques despite binding the protein.
- SPP1/osteopontin as master regulator: Gene expression analysis identified SPP1 as the most significantly upregulated factor induced by lecanemab treatment, with enriched expression around amyloid plaques in lecanemab-treated mice.
- Protective synapse preservation: Lecanemab-treated mice showed no significant loss of synapses near plaques, in marked contrast to untreated controls, preventing the neuronal damage cascade triggered by amyloid.
- Broad transcriptional reprogramming: Lecanemab induced coordinated upregulation of phagocytosis genes, lysosomal degradation pathways, metabolic reprogramming, and interferon response genes across microglial populations.
- Limited inflammatory activation: Despite robust amyloid clearance, lecanemab-treated microglia showed no broad pro-inflammatory cytokine release, suggesting a restrained immune activation that avoids toxic side effects.
- OPN-driven clearance in vitro: Human osteopontin at physiologic concentrations significantly enhanced amyloid clearance by microglia in cell culture, validating SPP1 as a direct functional driver.
Source: Nature Neuroscience (2026) | Albertini et al.
The Puzzle at the Heart of Lecanemab
Lecanemab’s clinical success in slowing Alzheimer’s cognitive decline set off urgent mechanistic questions. The antibody binds to soluble amyloid oligomers and insoluble plaques with high specificity, but binding alone doesn’t remove them from the brain. Something else had to bridge the gap between antibody engagement and actual plaque clearance.
The leading hypothesis pointed to Fc receptors—docking sites on immune cells that recognize the tail region of antibodies. But was this really the mechanism, and if so, which immune cells mattered most?
Microglia as the Critical Effector
To answer these questions, researchers used a clever experimental approach: xenotransplanted human microglia into the brains of transgenic Alzheimer’s mice. This let them watch real human immune cells—not mouse substitutes—respond to lecanemab in living brain tissue.
The results were unambiguous. Within 8 weeks of lecanemab treatment, amyloid plaque burden dropped sharply in regions with human microglia. But when the team used antibodies with crippled Fc regions, even though they still bound plaques perfectly, no clearance occurred. The immune-signaling tail was essential.
This finding overturned a competing hypothesis: that direct antibody-amyloid-complement interactions could drive clearance independently. Instead, microglia activation through Fc-receptor engagement emerged as the gating requirement for therapeutic benefit.
The SPP1/Osteopontin Discovery
To understand what lecanemab actually tells microglia to do, researchers performed single-cell RNA sequencing on human microglia exposed to lecanemab. The analysis revealed a remarkably coordinated gene expression shift. Among hundreds of differentially expressed genes, one stood out: SPP1, which encodes osteopontin, a secreted protein known for enhancing immune cell activation.
The fold-changes were dramatic. SPP1 expression spiked significantly in lecanemab-treated samples compared to controls. Spatially, when researchers mapped gene expression directly onto brain tissue around amyloid plaques, the SPP1 signal clustered intensely in the pink module—genes enriched for amyloid-proximity in lecanemab-treated brains.
To test causation, not just correlation, the team added purified human osteopontin to cultured microglia and amyloid. The result: osteopontin significantly enhanced plaque clearance in a dose-dependent manner. The effect was real and direct.
A Coordinated Microglial Awakening
Lecanemab’s effect wasn’t limited to a single gene. The transcriptional response involved tightly coordinated modules affecting multiple pathways. Gene set enrichment analysis revealed upregulation of phagocytosis machinery, lysosomal degradation enzymes, metabolic reprogramming (shifting energy availability toward the clearance task), and interferon response genes.
Notably, classical pro-inflammatory cytokine genes—the hallmark of dangerous microglial activation—showed no broad upregulation. This distinction matters clinically: lecanemab appeared to engage a targeted, functional immune response rather than a widespread inflammatory cascade.
The spatial transcriptomics data reinforced this picture. Lecanemab-treated brains showed enrichment of genes tied to antigen presentation, metabolic pathways, and protective immune programs—concentrated specifically around plaques. The microglia weren’t becoming globally inflamed; they were becoming locally competent.
Protection at the Synapse
One of the most striking findings concerned synaptic integrity. Untreated Alzheimer’s mice showed significant synapse loss around amyloid plaques—a hallmark of neurodegeneration. But in lecanemab-treated mice, synapses near plaques remained largely intact. This suggests that by rapidly clearing amyloid, lecanemab prevents the toxic local environment that normally triggers synaptic elimination.
Closer examination revealed why. Lecanemab reduced dystrophic neurites—the swollen, damaged neural processes that cluster around plaques in untreated disease. It also decreased LAMP1-positive dystrophic compartments, indicators of neuronal stress accumulation. By truncating this cascade early, the antibody prevented the downstream neuronal damage.
Implications for Future Anti-Amyloid Therapies
The study’s findings reshape how researchers should think about antibody engineering for Alzheimer’s. First, Fc-receptor engagement isn’t optional—it’s essential. Antibodies optimized purely for amyloid binding without attention to Fc function will fail therapeutically, even if they stick to target perfectly.
Second, microglia-mediated clearance appears to depend on specific transcriptional programs. SPP1/osteopontin emerged as a key brake on these programs; future therapies might explore whether enhancing osteopontin signaling, or compounds that mimic its effects, could amplify the microglial response without inducing systemic inflammation.
Third, the absence of broad inflammatory activation is instructive. Many researchers feared that engaging amyloid clearance would ignite neuroinflammation. Instead, lecanemab appears to activate a restrained, task-focused immune response—one that removes pathology without collateral damage.
Remaining Questions
Despite these insights, open questions remain. The study used xenotransplanted human microglia, which may behave differently than endogenous mouse microglia in certain contexts. The vascular pathology observed in some Alzheimer’s cases isn’t fully captured by brain-only models, leaving uncertainty about how peripheral immunity and blood-brain barrier function shape lecanemab’s in vivo efficacy.
Additionally, the temporal dynamics of Fc-receptor signaling—which receptors activate, in what sequence, at what intensity—remain incompletely understood. Future work dissecting FcγR versus complement receptor contributions could refine antibody design further.
[Insert image: spatial transcriptomics showing SPP1/osteopontin and phagocytosis gene enrichment around amyloid plaques in lecanemab-treated brains]
Citation: Albertini G, Zielonka M, Cuypers ML, et al. The Alzheimer’s therapeutic Lecanemab attenuates Aβ pathology by inducing an amyloid-clearing program in microglia. Nature Neuroscience. 2026;29:100–110. DOI: 10.1038/s41593-025-02125-8
Authors’ affiliations: Center for Brain and Disease Research, Flanders Institute of Biotechnology (VIB), Leuven, Belgium; Department of Neurosciences and Leuven Brain Institute (LBI), KU Leuven, Belgium; Laboratory for Therapeutic and Diagnostic Antibodies, Department of Pharmaceutical and Pharmacological Sciences, KU Leuven, Belgium; Single Cell Core, Flanders Institute of Biotechnology (VIB), Belgium; Center for Molecular Neurology, Flanders Institute for Biotechnology (VIB), Antwerp, Belgium; Department of Biomedical Sciences, University of Antwerp, Belgium; UK Dementia Research Institute, University College London, London, UK; Department of Human Sciences, KU Leuven, Belgium.