TL;DR: A 2026 mouse study in Proceedings of the National Academy of Sciences found that deleting or inhibiting PTP1B improved memory behavior, reduced amyloid-beta burden, and pushed microglia toward an amyloid-clearing state.
Key Findings
- Memory behavior improved: APP/PS1 mice lacking PTP1B performed better in novel object recognition and Morris water maze tests; the PTP1B inhibitor DPM-1003 produced a similar behavioral rescue.
- Amyloid staining fell 26-33%: PTP1B deletion reduced hippocampal Thioflavin S plaque area by 33% and 6E10-positive amyloid area by 27%; DPM-1003 reduced those measures by 28% and 26%.
- Soluble amyloid-beta 42 dropped 48%: Genetic deletion reduced soluble A-beta 42 by 48% and insoluble A-beta 42 by 35%; DPM-1003 reduced them by 28% and 30%.
- Microglia became more phagocytic: PTP1B-deficient microglia showed higher amyloid engulfment, more CD68-positive phagolysosome signal, and a transcriptomic shift toward disease-associated microglia.
- SYK was the mechanistic brake point: The study identified SYK as a direct PTP1B substrate, and blocking SYK reduced the enhanced phagocytosis and metabolic activation.
Source: PNAS (2026) | Cen et al.
PTP1B inhibition is not the usual starting point for an Alzheimer’s story. The field often begins with amyloid plaques, tau tangles, or antibody drugs.
This paper starts with a phosphatase, an enzyme that removes phosphate groups from signaling proteins, and asks whether taking that brake off immune cells can help the brain clear amyloid-beta.
The answer, in mice, was yes.
In APP/PS1 mice, a widely used amyloid-model of Alzheimer’s disease, deleting PTP1B or blocking it with the inhibitor DPM-1003 improved memory behavior and reduced amyloid-beta burden.
The strongest part of the paper is not just that amyloid went down.
It is that the authors mapped a plausible cellular route: PTP1B restrained SYK signaling in microglia, and removing that restraint made the cells more metabolically active and more phagocytic.
PTP1B Deletion Improved Memory Tests in APP/PS1 Mice
The first test was whether PTP1B changed whole-animal behavior.
The researchers crossed PTP1B knockout mice with APP/PS1 mice, then tested learning and memory when the animals were 12-13 months old, an age when this model has amyloid pathology and cognitive deficits.
The mouse data point in 3 linked directions:
- Behavior: PTP1B deletion or inhibition improved memory-task performance in APP/PS1 mice.
- Amyloid: multiple amyloid-beta measures decreased after genetic or drug-based PTP1B reduction.
- Microglia: immune cells shifted toward a more phagocytic amyloid-clearing state.
Human translation still has 3 constraints:
- Model limit: APP/PS1 mice do not reproduce the full biology of human Alzheimer’s disease.
- Cell-type limit: global deletion and systemic inhibition can affect neurons, glia, and peripheral tissues.
- Safety limit: PTP1B also regulates metabolism and immune signaling outside the brain.
In the novel object recognition test, APP/PS1 mice lacking PTP1B spent more time exploring the new object than APP/PS1 controls.
In the Morris water maze, PTP1B deletion shortened escape latency during learning and increased both platform crossings and time in the target quadrant during the probe test.
Swimming speed and open-field behavior did not explain the effect, which helps separate memory performance from motor or anxiety-like behavior.
The drug experiment made the result more relevant. Starting at 11 months of age, mice received DPM-1003, an allosteric PTP1B inhibitor, for 5 weeks.
Treated APP/PS1 mice again improved in novel object recognition and water maze measures, suggesting the effect was not limited to animals missing PTP1B from birth.
Amyloid-Beta Burden Fell Across Multiple Measures
The behavioral rescue would be less convincing if amyloid pathology stayed unchanged. It did not.
In hippocampal tissue, PTP1B deletion reduced Thioflavin S-positive plaque area by 33% and 6E10-positive amyloid area by 27% . DPM-1003 treatment produced a similar pattern, reducing Thioflavin S-positive area by 28% and 6E10-positive area by 26% .
The biochemical measurements pointed the same way.
In cortical tissue, PTP1B deletion reduced soluble A-beta 42 by 48% and insoluble A-beta 42 by 35% . DPM-1003 reduced soluble A-beta 42 by 28% and insoluble A-beta 42 by 30% .
The authors also checked whether this was simply a production effect.
PTP1B deletion did not change full-length APP, APP C-terminal fragments, BACE1, or presenilin 1 in the way one would expect if amyloid production had been reduced upstream.
That pushed the interpretation toward clearance: the brain appeared to be removing more amyloid-beta rather than making less of it.
Microglia Shifted Toward an Amyloid-Clearing State
Microglia are the brain’s resident immune cells, and one of their jobs is to engulf debris and protein aggregates. In Alzheimer’s disease, that function can become inadequate or dysregulated.
Cen et al. found that Ptpn1, the gene encoding PTP1B, was highly expressed in brain immune cells, including microglia, which made those cells a natural place to look.
Single-cell RNA sequencing in 13-month-old female APP/PS1 mice showed that PTP1B deletion shifted microglia toward a more activated state.
Disease-associated microglia increased from 44.9% in APP/PS1 controls to 51.7% in APP/PS1 mice lacking PTP1B.
Genes linked to lysosomes, cathepsins, antigen presentation, Trem2, Axl, Cd9, and phagocytosis were upregulated.
The imaging and cell assays supported the transcriptomics. PTP1B-deficient microglia showed higher baseline phagocytic activity, responded more strongly to amyloid-beta oligomers, and engulfed more amyloid-beta in brain slices.
Amyloid internalization within CD68-positive phagolysosomes was more than doubled without PTP1B, which is exactly the direction the clearance hypothesis would predict.
SYK Connected the Immune Signal to the Amyloid Result
The mechanistic center of the paper is SYK, a kinase that helps coordinate microglial responses to amyloid pathology. PTP1B-deficient microglia showed increased SYK phosphorylation after amyloid-beta oligomer stimulation.
They also showed stronger PI3K-AKT-mTOR signaling, higher glycolytic output, and greater mitochondrial respiration, all consistent with a cell shifting into an energy-demanding active state.
The authors then blocked SYK with BAY61-3606. That reduced the enhanced phagocytosis, AKT-mTOR signaling, lactate production, and oxygen consumption seen in PTP1B-deficient microglia.
In other words, SYK was not just a marker that changed alongside the phenotype. It was required for much of the boosted microglial response.
The study went one step deeper by showing that SYK is a direct substrate of PTP1B. Substrate-trapping experiments and mutation of SYK activation-loop residues Y525 and Y526 supported a direct enzyme-substrate relationship.
The simple model is that PTP1B normally dephosphorylates SYK, keeping microglial activation in check. Remove or inhibit PTP1B, and SYK signaling stays more active.
PTP1B Inhibition Still Needs Human Alzheimer’s Testing
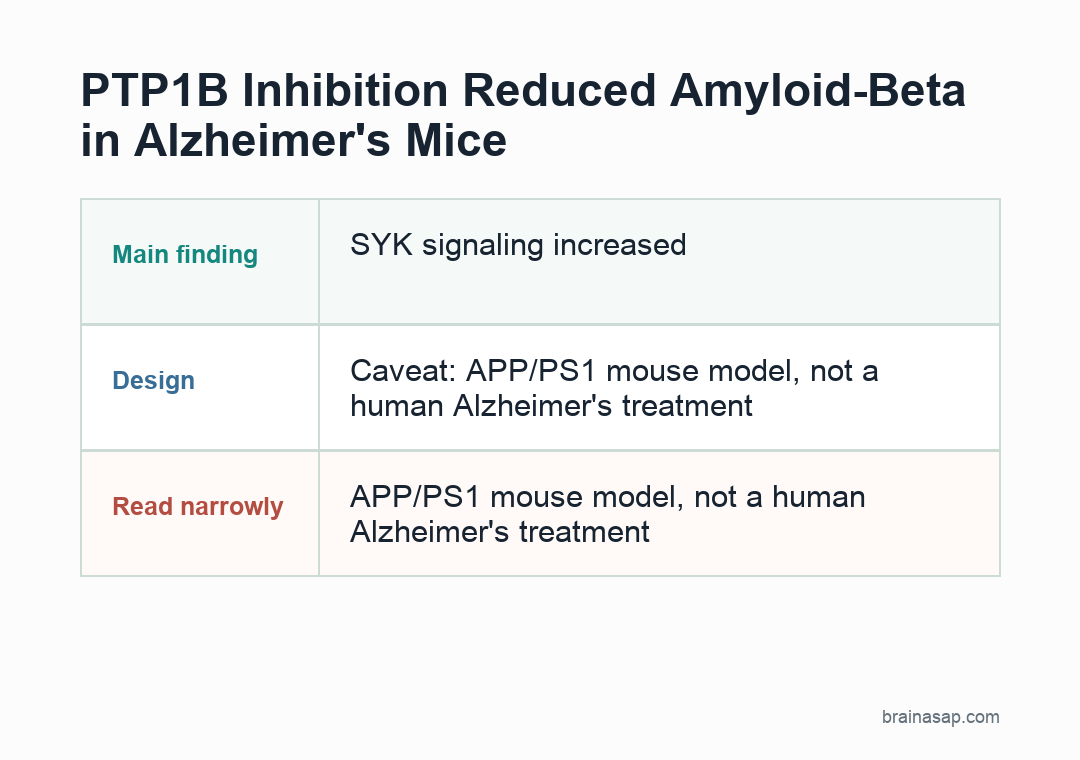
This is not evidence that a PTP1B inhibitor treats Alzheimer’s disease in people. The central disease data came from APP/PS1 mice, a model built around amyloid pathology.
Mouse amyloid models are useful for mechanism, but they do not reproduce the full human disease, especially tau pathology, vascular injury, aging heterogeneity, and years of clinical progression.
- Model limit: APP/PS1 mice are built to develop amyloid pathology, so the result needs testing in additional Alzheimer’s models.
- Cell-type limit: Global PTP1B deletion and systemic DPM-1003 treatment cannot fully separate microglial effects from neuronal or peripheral effects.
- Translation limit: A human therapy would need the right brain exposure, dose window, and immune-metabolic safety profile.
Cell type still needs separation.
The study highlights a new microglial role for PTP1B, but global deletion and systemic DPM-1003 treatment could affect neurons and other cells too.
The authors explicitly note that microglia-specific and neuron-specific PTP1B ablation will be needed to separate those contributions.
Safety remains central.
PTP1B regulates metabolism and immune signaling across tissues, which is part of why the target is biologically plausible and therapeutically complicated.
A useful Alzheimer’s therapy would need brain-relevant exposure, enough microglial effect, and acceptable systemic immune and metabolic safety.
Why This Target Is Worth Watching
The paper is strongest as a mechanism-and-target study.
It links a druggable phosphatase to microglial amyloid clearance, identifies SYK as a direct signaling node, and shows that both genetic and pharmacological PTP1B targeting moved the same major outcomes in the right direction.
That combination is more persuasive than a single behavioral improvement or a single plaque measurement. The result connects behavior, amyloid burden, microglial state, metabolism, and enzyme-substrate chemistry into one testable path.
If future studies can reproduce the effect in additional models and define the safest cell type and dose window, PTP1B could become a serious candidate in the broader push to make microglia better amyloid-clearing cells.
Citation: DOI: 10.1073/pnas.2521944123. Cen et al. PTP1B inhibition promotes microglial phagocytosis in Alzheimer’s disease models by enhancing SYK signaling. Proceedings of the National Academy of Sciences. 2026;123:e2521944123.
Study Design: Mouse genetic-deletion and inhibitor study of PTP1B, microglia, SYK signaling, and amyloid burden.
Sample/Model: APP/PS1 Alzheimer’s model mice with PTP1B deletion or DPM-1003 inhibition.
Key Statistic: PTP1B reduction improved memory behavior and reduced multiple amyloid-beta measures while increasing microglial phagocytosis.
Caveat: APP/PS1 mouse findings need human validation and safety testing before clinical Alzheimer’s claims.