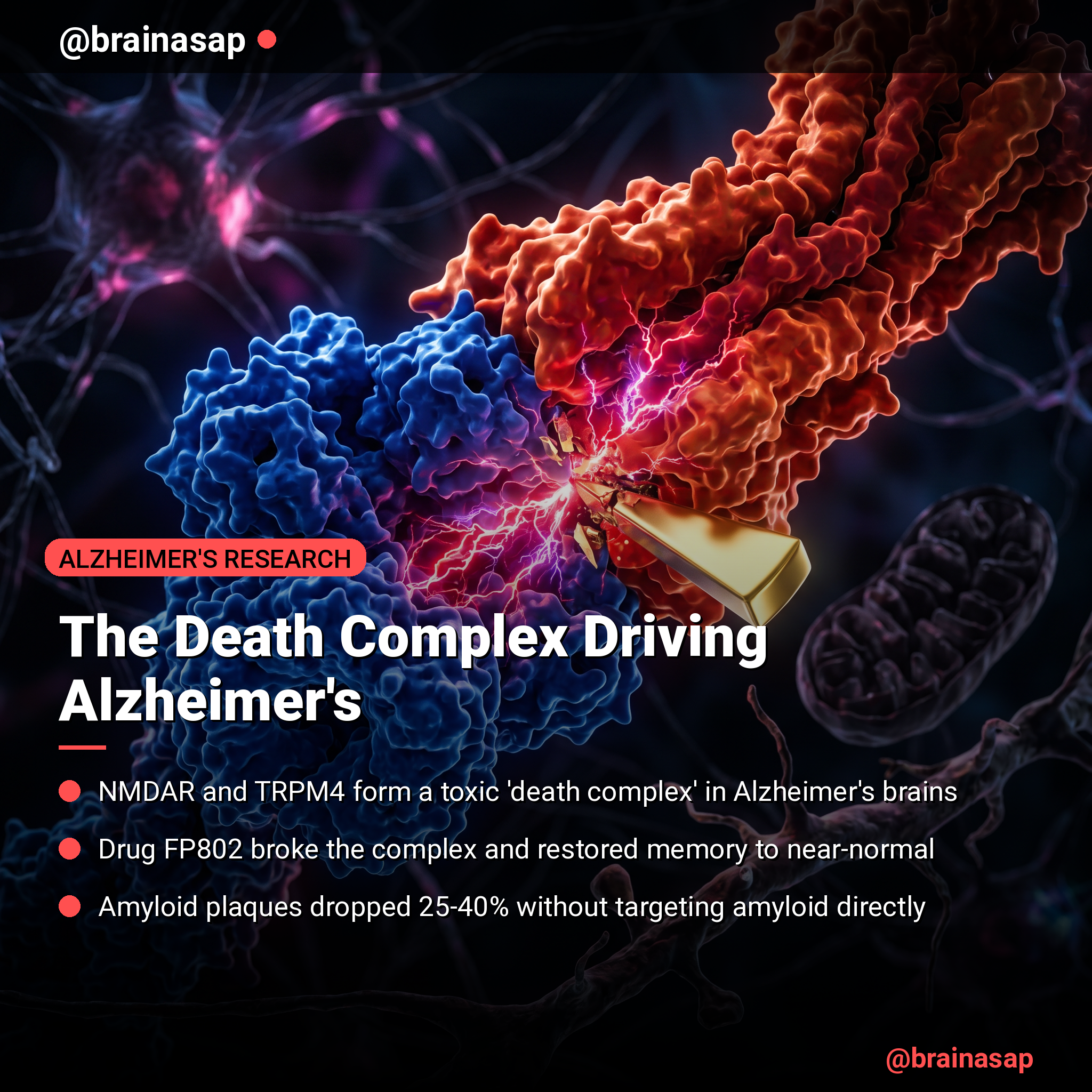
The Death Complex Fueling Alzheimer’s (And How to Stop It)
TL;DR: Researchers discovered a toxic interaction between two brain proteins—NMDAR and TRPM4—that drives neurodegeneration in Alzheimer’s disease, and a small molecule called FP802 that blocks this “death complex” and prevents cognitive decline in mice.
Alzheimer’s disease devastates the brain through multiple pathways, but the exact triggering mechanisms remain frustratingly unclear. A new study in Molecular Psychiatry identifies a previously unknown culprit: a specific protein-protein interaction that acts like a death switch for neurons, and demonstrates a drug that can flip it off.
Key Findings
- The NMDAR/TRPM4 death complex forms in Alzheimer’s brains: In 5xFAD mice (a genetic model of the disease), the researchers detected increased interaction between two proteins—the NMDA receptor (NMDAR) and a calcium channel called TRPM4—that do not normally bind together at high levels. Blocking this complex with FP802 reduced GluN2A-TRPM4 co-interactions by 75-80%.
- FP802 restores spatial memory and learning: In Morris water maze tests (a standard memory assessment), vehicle-treated 5xFAD mice showed impaired memory, but FP802-treated mice performed as well as healthy controls, achieving 70-80% accuracy in probe tests compared to 40% in untreated Alzheimer’s mice.
- Dendritic spines and synapses are preserved: The drug prevented loss of dendritic complexity and spine density in hippocampal CA1 neurons. Untreated 5xFAD mice showed a 27-33% reduction in dendritic length; FP802 restored it, maintaining neural architecture necessary for memory.
- Mitochondrial function improves dramatically: Electron microscopy revealed that untreated 5xFAD mice had severely swollen, dysfunctional mitochondria. FP802 treatment shifted the distribution from 27% swollen mitochondria back to normal or near-normal morphology, restoring energy production for neurons.
- Synaptic integrity is restored: Examination of synapses showed FP802 prevented loss of excitatory and inhibitory synapses. The drug reduced spine pathologies and maintained synapse length and thickness, suggesting neuronal communication circuits remain functional.
- Amyloid plaque burden is reduced by 25-40%: Immunohistochemistry revealed that FP802 treatment significantly lowered amyloid-beta (Aβ) accumulation in both the hippocampus and cortex compared to vehicle-treated Alzheimer’s mice, indicating reduced pathological protein aggregation.
Source: Molecular Psychiatry (2026) | Yan et al.
A Deadly Partnership: The NMDAR/TRPM4 Complex
For decades, Alzheimer’s research has focused on amyloid-beta plaques and tau tangles as the primary drivers of neurodegeneration. But the brain is more complex than that. Amyloid and tau may initiate the disease, but they activate multiple downstream pathways—and one of those pathways has remained in shadow until now.
The NMDAR/TRPM4 death complex represents a direct conversation between two proteins that normally operate independently. The NMDA receptor is a glutamate-sensing ion channel critical for memory formation. TRPM4 is a calcium channel involved in cellular stress responses. When amyloid-beta and other pathological signals activate them simultaneously, they bind together and trigger a cascade of cell death.
The discovery is significant because it offers a mechanistic target—not amyloid itself, but a specific molecular handshake downstream of it. This is why blocking this single interaction can have such profound effects on multiple levels of neurodegeneration at once.
FP802: A TwinIF Inhibitor That Breaks the Complex
The researchers used a clever pharmacological strategy: they deployed FP802, a small molecule from a new drug class called TwinIF (Twin Interface) inhibitors. These compounds are designed to disrupt protein-protein interactions by binding to the interface where two proteins meet.
Unlike classical NMDAR blockers—which shut down the entire channel and cause severe side effects—FP802 is surgical in its precision. It specifically disrupts the NMDAR/TRPM4 interaction while leaving normal NMDAR signaling intact. This distinction is critical. It allows neuroprotection without the cognitive fog and dissociative effects that plagued earlier NMDAR antagonists.
The team tested two doses: 10 mg/kg and 40 mg/kg daily. Both were effective, suggesting a wide therapeutic window. Remarkably, FP802 showed no adverse effects on liver, kidney, or heart function in treated mice, indicating good safety.
Cognitive Rescue Across Three Memory Tests
The cognitive effects were striking. In the Morris water maze—the gold standard for rodent spatial memory—untreated 5xFAD mice showed progressive decline over six days of training, reaching only 30-40% success in finding the hidden platform. FP802-treated mice learned steadily and achieved 70-80% accuracy by day 6, indistinguishable from healthy controls.
The researchers then tested other forms of memory. In a novel object recognition task (a test of recognition memory), FP802 prevented the cognitive deficits that plague untreated Alzheimer’s mice. In contextual fear conditioning (associative memory), FP802 restored the ability to link context with fear—a memory function that depends critically on the hippocampus.
These are not subtle improvements. The drug reversed cognitive decline across three independent domains of memory, suggesting broad protection of hippocampal function.
Preserving the Neuronal Architecture
But cognition is only the behavioral readout. Underneath, the real victory lies in what the drug preserves at the cellular level. Using confocal microscopy and three-dimensional reconstruction, the researchers traced the complete dendritic trees of individual CA1 neurons from the hippocampus.
Untreated 5xFAD mice showed catastrophic structural loss. Total dendritic length was reduced by 27-33%, and dendritic spines—the tiny protrusions that receive synaptic input—were dramatically thinned. FP802 treatment arrested this decay. Dendritic length was restored to near-normal levels, and spine density and spine head size remained intact.
Zooming in further, the researchers counted individual synapses using electron microscopy. Untreated Alzheimer’s mice showed a 50-60% loss of both excitatory and inhibitory synapses. FP802 prevented this collapse. Synapses remained present, robust, and correctly structured—with intact postsynaptic density (PSD) scaffolds that are essential for signal transmission.
Restoring Mitochondrial Energy
One of the least discussed but most critical aspects of Alzheimer’s pathology is mitochondrial dysfunction. These cellular power plants normally have a defined shape (roughly elliptical); in neurodegeneration, they become swollen and fragmented, unable to generate ATP.
The researchers used transmission electron microscopy to categorize mitochondrial morphology across four classes: normal, vesicular, vesicular-swollen, and severely swollen. In healthy control mice, approximately 54-62% of mitochondria were normal. In untreated 5xFAD mice, this plummeted to 17-27%, with the majority swollen and dysfunctional.
FP802 treatment shifted the distribution back toward normal. The proportion of normal mitochondria increased dramatically, and severe swelling was prevented. This restoration of mitochondrial structure correlates directly with improved cognitive function—neurons with functional power supplies can sustain synaptic transmission and maintain memory.
Dampening Amyloid Accumulation
Finally, the researchers examined whether FP802 influences amyloid-beta pathology itself. Using Congo Red staining to visualize amyloid plaques in hippocampus and cortex, they found that untreated 5xFAD mice accumulated extensive plaque burden. FP802 treatment reduced amyloid load by 25-40% in both brain regions.
This is noteworthy because FP802 is not an anti-amyloid drug—it doesn’t target amyloid production or clearance. Yet by blocking the NMDAR/TRPM4 death signal, it appears to reduce the pathological stress signals that promote amyloid aggregation. In other words, disrupting the death complex may reduce the oxidative stress and inflammatory environment that fuels amyloid accumulation.
[Insert image: diagram showing NMDAR/TRPM4 interaction and FP802 TwinIF inhibitor blockade mechanism in Alzheimer’s neurons]
What Makes This Different From NMDAR Blockers
It’s important to understand why this approach succeeds where decades of NMDAR antagonists failed. Classical NMDAR blockers like memantine inhibit the entire channel—they block all glutamate signaling through NMDAR, which is necessary for learning, memory consolidation, and synaptic plasticity.
FP802 takes a different tack. It allows normal NMDAR signaling to continue. It only disrupts the pathological NMDAR/TRPM4 interaction that emerges under disease conditions. This explains why the drug can rescue memory without causing the cognitive side effects that derailed earlier NMDAR-targeted therapies.
The TwinIF inhibitor approach also applies beyond Alzheimer’s. The researchers note that NMDAR/TRPM4 interactions drive toxicity in other neurodegenerative diseases, including amyotrophic lateral sclerosis (ALS), in which FP802 has already shown promise in preclinical models.
Limitations and Next Steps
This is a mouse study, and translation to humans is never guaranteed. The 5xFAD model represents early-to-mid-stage Alzheimer’s pathology; it’s unclear whether FP802 would benefit patients at more advanced disease stages. Additionally, the study focused on cognitive and structural endpoints; longer-term survival and lifespan effects were not measured.
The mechanism of how FP802 reduces amyloid plaque burden also warrants further investigation. If the drug’s primary target is the NMDAR/TRPM4 interaction, the reduction in amyloid may be an indirect consequence of reduced neuronal stress.
Despite these caveats, the findings represent a genuine conceptual advance. The identification of the NMDAR/TRPM4 death complex as a major driver of disease progression, combined with demonstration that disrupting it can reverse multiple pathological features simultaneously, opens a new therapeutic avenue. FP802 is advancing toward human trials, and if it maintains its safety profile and cognitive benefits in patients, it could represent a meaningful shift in how we treat Alzheimer’s disease.
Citation: Yan J, Yang X, Li G, Ramirez OA, Hagenston AM, Chen ZY, et al. The NMDAR/TRPM4 death complex is a major promoter of disease progression in the 5xFAD mouse model of Alzheimer’s disease. Molecular Psychiatry. 2026;31:635-648. DOI: 10.1038/s41380-025-03143-5
Authors’ affiliations: Department of Neurobiology, Interdisciplinary Center for Neuroscience (IZN), Heidelberg University, Germany; FundaMental Pharma GmbH, Heidelberg, Germany; Department of Anatomy and Neurobiology, Shandong Key Laboratory of Mental Disorders and Intelligent Control, School of Basic Medical Sciences, Cheeloo College of Medicine, Shandong University, Jinan, Shandong, P.R. China; Research Center for Basic Medical Sciences, Qilu Hospital of Shandong University, Jinan, Shandong, P.R. China; State Key Laboratory for Innovation of Luobing Theory, Jinan, Shandong, P.R. China; Network Aging Research, Heidelberg University, Germany.